Command Palette
Search for a command to run...
Selected for ICML 2025, Tsinghua University/Renmin University Proposed UniSim, a Unified Biomolecular Dynamics Simulator

The group of Professor Liu Yang from Tsinghua University and the group of Professor Huang Wenbing from the Gaoling School of Artificial Intelligence at Renmin University of China jointly proposed a unified biomolecular time-coarsened dynamics simulator UniSim.This method obtains a unified all-atom representation model through denoising + force field hybrid pre-training on a large amount of 3D molecular structure data, learns the transfer vector field of molecules at a long time step based on a stochastic interpolant generative framework, and introduces a force-guided core to quickly adapt to different chemical environments. UniSim is the first to realize a unified time-coarsened dynamics simulation framework across molecular types (small molecules, peptides, proteins) and chemical environments.It has promoted the practical application of deep learning in the field of molecular simulation.
The related results were selected for ICML 2025 under the title "UniSim: A Unified Simulator for Time-Coarsened Dynamics of Biomolecules".

Paper address:
More AI frontier papers:
https://go.hyper.ai/UuE1o
Why do we need a unified temporal coarsening simulator?
The researchers believe that in the field of molecular dynamics simulation, it is reasonable and necessary to build a unified time-coarsening simulator.On the one hand, a unified modeling framework is the basis for collaborative simulation across molecular systems.For example, when simulating complex systems such as protein-ligand interactions, proteins and small molecules often coexist in the same physical environment. If the model is only applicable to a certain type of molecule, it will be difficult to accurately restore the coupling behavior between the two at the all-atomic scale. Therefore, simulators with unified representation capabilities can simultaneously handle cross-type molecules within the same model framework, providing a solid foundation for modeling multi-molecular complexes.
On the other hand, a unified model helps to integrate the structural and dynamic data of different types of molecules, thereby improving the generalization and transfer capabilities of the model.Currently available molecular trajectory data are highly scarce and unevenly distributed, and various types of data such as proteins, peptides, and small molecules have their own strengths. If they can all participate in pre-training and learning in the same model, it will significantly enhance the model's overall understanding of atomic-level structures and enable it to have stronger cross-molecular domain migration capabilities.
at the same time,Introducing time coarsening simulation is also a core way to improve simulation efficiency.Traditional molecular dynamics simulations rely on extremely small time steps (such as femtoseconds) to advance step by step, which is computationally expensive and difficult to cover long-term behaviors such as protein folding. The time coarsening method directly learns the mapping relationship from the current state to the future state. Under the premise of maintaining physical consistency, it can quickly generate trajectories at a time scale much larger than the traditional step size, greatly improving the simulation efficiency and making it possible to perform long-term simulations in a practical time.
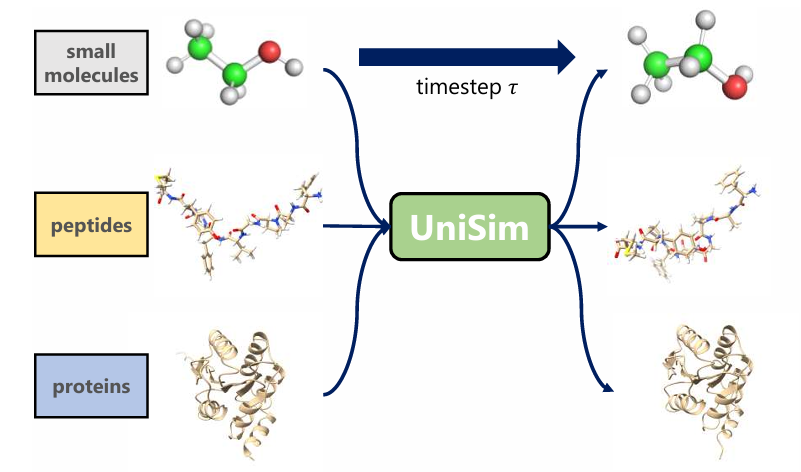
Unified representation: solving the problem of characterizing multi-scale and multi-type molecules
Although unified all-atom representation models are the cornerstone of dynamics simulations across molecular species,However, the implementation of such a model still faces the following technical challenges:
* First, molecular systems range from small organic molecules of dozens of atoms to protein macromolecules of thousands of atoms, with huge differences in scale and complex and diverse structures.If all atoms are used for training directly, the model will have different attention mechanisms for different types of molecules, thereby inhibiting the cross-transfer ability of the model.
* Secondly, the basis for achieving a unified all-atom representation is to use a unified atomic-level vocabulary.An intuitive approach is to directly use the periodic table as a vocabulary for embedding representation. However, this approach ignores the regular units that exist in large numbers in peptides and proteins, such as substructures such as natural amino acids, resulting in poor performance on protein type data.
* Finally, in order to fully learn the representation of molecules in different states, a large amount of steady-state and non-steady-state molecular 3D structure data will be included in the pre-training dataset.The common paradigm for pre-training non-stable molecules is to learn the forces acting on atoms. However, different datasets use different force field parameters when calculating atomic force fields, and there is a misalignment of label data.
In order to achieve unified modeling, UniSim introduces three key technologies to solve the above problems:
* Gradient-environment subgraph: reasonably balancing the molecular scale
In the data preprocessing stage, the 3D structure data of large molecules (with more than 1,000 atoms) will be segmented.min < rmax During preprocessing, any atom in the molecule will be randomly selected, and the atom will be taken as the center of the sphere.min and rmax Make a sphere with radiusThe atoms contained in the small ball are regarded as the gradient subgraph, and the atoms contained in the large ball are regarded as the environment subgraph.Based on the physical prior that the interatomic force generally decays exponentially with distance, when rmax– rmin When properly selected, the interaction between atoms outside the environment subgraph in the original molecule and atoms in the gradient subgraph will be negligible. Therefore, during training, the environment subgraph will be used instead of the original molecule as input, and only the gradient subgraph will be involved in the loss function calculation, thereby reasonably balancing the scale of molecular structure data and improving the model's cross-transfer capability.
* Atom embedding extension: Get more refined atomic representation
This study is based on the periodic table of elements.Introducing multiple learnable discrete embedding representations for the same element as an extended vocabulary,It is used to capture the regular substructure where atoms are located. Based on a simple graph neural network, UniSim will integrate the neighborhood information of each atom, obtain the probability of each embedded representation in the extended vocabulary corresponding to the atom, and obtain the extended embedded representation of the atom through weighted summation.This representation balances atomic-level accuracy with regular substructures within specific molecular species, resulting in efficient and detailed atomic representation.
* Multi-head hybrid pre-training: hybrid learning of data with different molecular states and label distributions
UniSim uses the following method to jointly learn steady-state and non-steady-state molecular structures: for steady-state data, the article uses the denoising pretraining paradigm to denoise the noisy data to learn atomic representation; for non-steady-state data, the model will directly learn the conservative force field, and different force field parameters correspond to different output heads, thereby avoiding the errors introduced by different label distributions.
The article uses TorchMD-NET as the basic graph neural network model that satisfies SO(3) equivariance. Based on the above key pre-training techniques,Pre-training on large-scale multi-source 3D molecular data was completed, and the effective construction of a unified atomic representation model was achieved.
Vector field models: learning long-time state transitions from trajectories
Traditional molecular dynamics simulations are limited by integration steps of a few femtoseconds, making it difficult to efficiently sample long-term behaviors such as protein folding. UniSim adopts a stochastic interpolant framework and connects a geometric vector perceptron as a vector field model after a pre-trained all-atom representation model.The model achieves end-to-end time-coarsened dynamics modeling by learning the transfer vector field between molecular states at long time steps.
During training, pairs of molecular conformations separated by a given time step in the real dynamics trajectory are selected as training samples, random perturbations are introduced on the interpolation path, and the velocity field (velocity) and denoiser (denoiser) are jointly learned to achieve trajectory generation in continuous time. Compared with traditional numerical integration, UniSim can significantly improve simulation efficiency and break through the bottleneck of traditional simulation in time scale.
Force-guided nuclei: Rapid adaptation to complex chemical environments
Molecular dynamics under different solvent, temperature and pressure conditions have different potential energy surfaces, which greatly affect the distribution of generated conformations.To this end, UniSim introduces a force guidance kernel to define a virtual intermediate force field on the random difference framework to guide trajectory sampling.This intermediate force field is equivalent to the real MD force field at both ends of the generation path (i.e., the initial state and the final state), and is designed to be highly consistent with the physical priors, so that the generated conformation is more consistent with the Boltzmann distribution under the target force field.
By fitting the intermediate force field, UniSim does not need to modify the parameters of the pre-trained model and the vector field model.Only a pluggable force-guiding kernel needs to be learned for the target force field to efficiently adapt to new chemical environments.Effectively enhances the generalization and migration capabilities of the model.
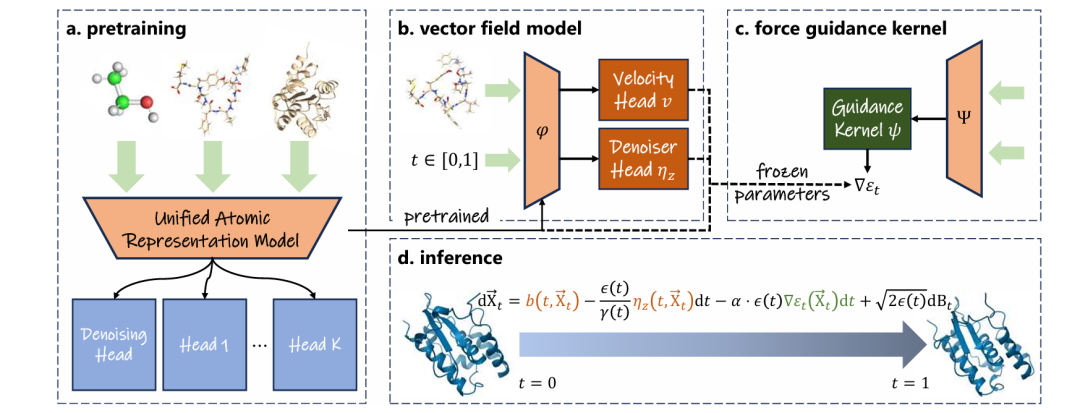
Experimental verification: multiple molecule types
In order to verify the versatility of UniSim on different molecular types,The researchers systematically evaluated data from multiple molecular types in a forward simulation task, including three types of molecules: small molecules, peptides, and proteins.By comparing with the deep learning model in the field that also performs time-coarsened dynamics simulation, the experiment aims to explore whether the unified atomic representation can help improve the model's understanding of molecular states and cross-modal generalization capabilities, and how the involvement of force-guided nuclei affects the model's performance in key indicators such as the rationality of conformations generated under the target force field and distribution similarity.
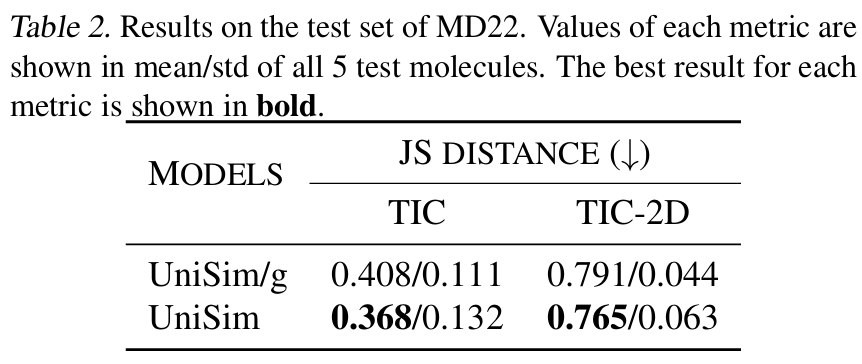
The results show that UniSim has achieved comprehensive superiority in all molecular types.It performs well in distribution similarity and has a significant improvement in the key conformational rationality indicator (Val-CA). It should be noted that in the forward simulation generation task, each conformation in the trajectory is generated by autoregression, which has a huge cumulative error, so it is quite difficult to improve the rationality of the conformation.
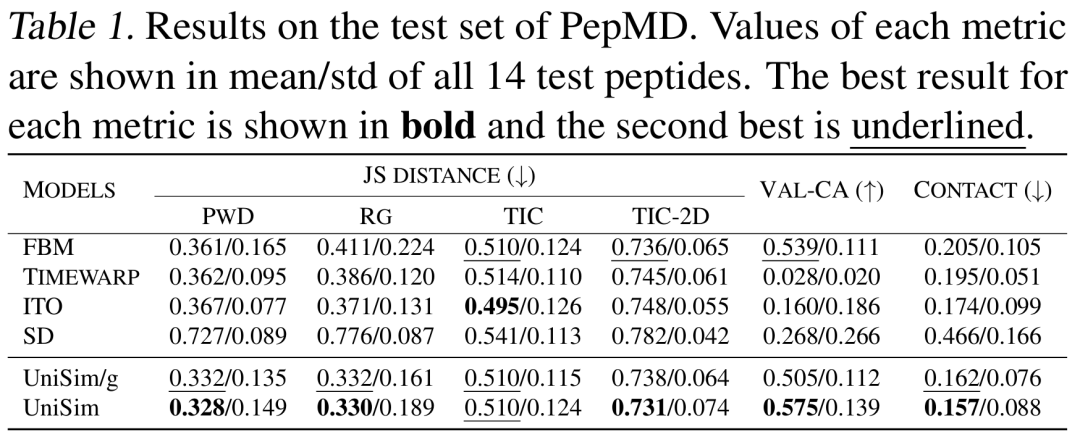
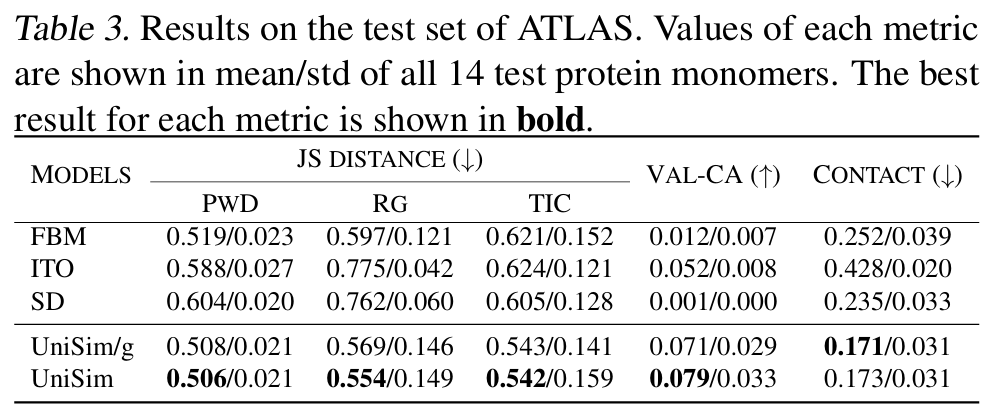
In the forward simulation task of peptides and proteins,Compared with existing methods such as FBM, ITO, and SD, UniSim is ahead in indicators such as distribution similarity (TIC-2D), structural rationality (VAL-CA), and contact map error (CONTACT). In particular, after the introduction of the force-guided core, UniSim maintains the original level in indicators such as distribution similarity, but has significantly improved in key conformational rationality indicators. At the same time, in complex protein systems, UniSim can jump energy barriers and cover multiple metastable states through only hundreds of steps of forward simulation, opening up new directions for efficient simulation of large biomolecules.
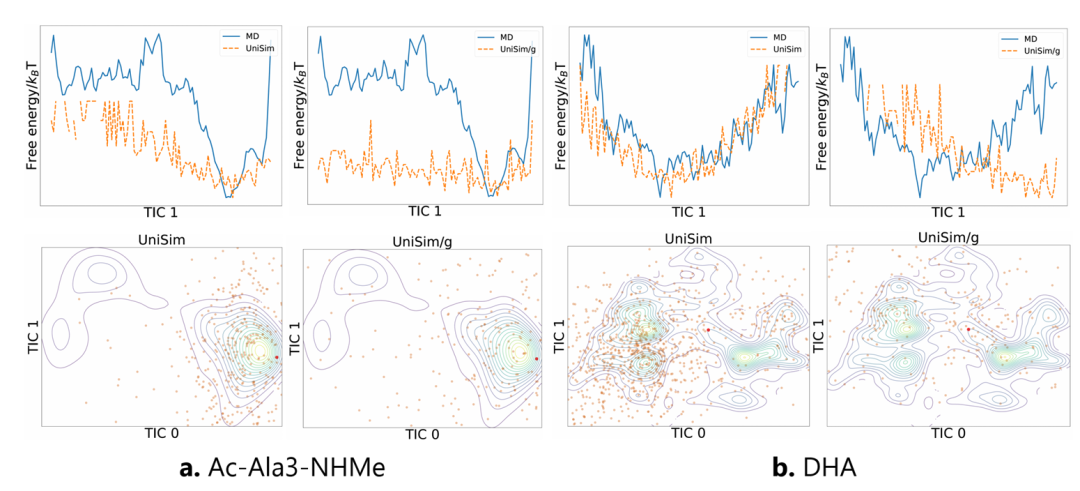
Alanine-Dipeptide Case Study
Furthermore, to explore the stability of UniSim in long-term molecular dynamics simulations, the researchers fine-tuned the model on the classic system alanine-dipeptide and performed long-time simulations of 100,000 steps.By comparing with MD results, UniSim successfully reproduced 5 known key metastable states.The free energy landscape of alanine dipeptide in the dynamic process was accurately restored, fully verifying the stability and physical consistency of the model under long-term simulation.
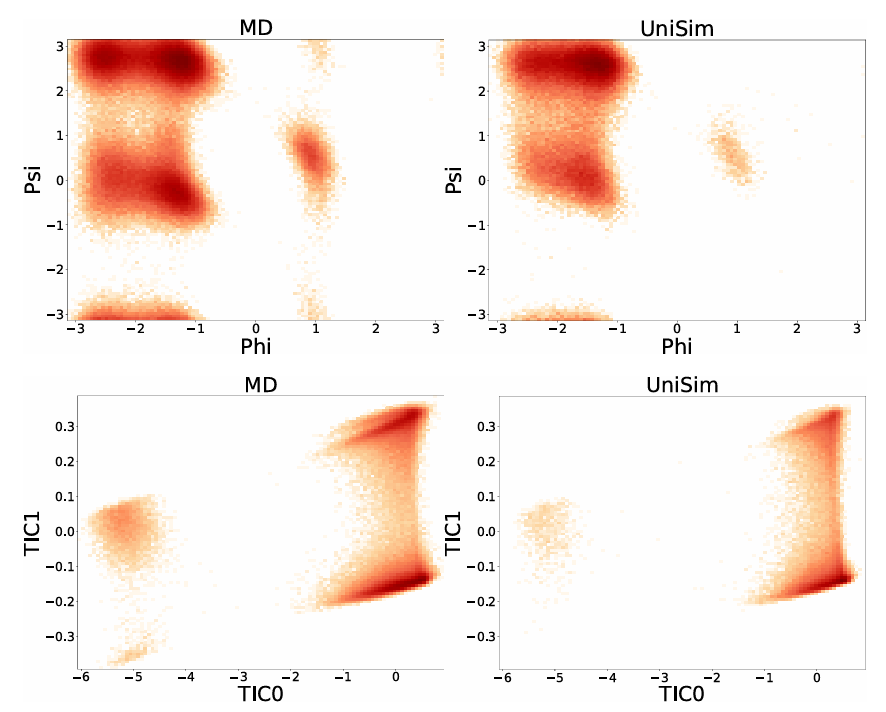
Outlook
UniSim is the first framework to achieve unified temporal coarse-grained dynamics simulation across molecular types and chemical environments.This provides a feasible path for the widespread application of deep learning in drug discovery, protein design and other scenarios. The researchers also pointed out that the following directions can be further explored in the future:
* A more efficient cross-modal conformation optimization mechanism to improve the effectiveness of generated samples;
* Trajectory modeling on longer time scales to reveal complex biophysical mechanisms;
* Explore the dynamic mechanisms in complex systems, focusing on intermolecular interactions.








