Command Palette
Search for a command to run...
8k Long Sequence Modeling, Protein Language Model Prot42 Can Generate High Affinity Binders Using Only the Target Protein Sequence
Protein binders (such as antibodies and inhibitory peptides) play an irreplaceable role in key scenarios such as disease diagnosis, imaging analysis, and targeted drug delivery.Traditionally, the development of highly specific protein binders has relied heavily on experimental techniques such as phage display and directed evolution.However, such methods generally face the challenges of huge resource consumption and lengthy R&D cycles, and are limited by the inherent bottleneck of the complexity of protein sequence combinations.
With the development of artificial intelligence, protein language models (PLMs) have become an important tool for understanding the relationship between protein sequences and functions. For protein binder design, PLMs can directly design ligand proteins or antibody fragments with high binding affinity based on the target protein sequence based on the generation ability of language models. However, it also faces challenges, such as the lack of PLMs with both long-context modeling capabilities and true generation capabilities, especially in the design of complex binding interfaces and long protein binders. There is a significant technical gap.
Based on this, a joint research team from the Inception AI Institute in Abu Dhabi, UAE and Cerebras Systems in Silicon Valley, USA,The first PLMs family, Prot42, which relies only on protein sequence information and does not require three-dimensional structure input was proposed.This model exploits the generative power of autoregression and decoder-only architecture.Enables generation of high-affinity protein binders and sequence-specific DNA-binding proteins in the absence of structural information.Prot42 performed well in PEER benchmark, protein binder generation, and DNA sequence-specific binder generation experiments.
The related research is titled "Prot42: a Novel Family of Protein Language Models for Target-aware Protein Binder Generation" and has been published as a preprint on arXiv.
Research highlights* Prot42 uses a progressive context expansion training strategy, which gradually expands from the initial 1,024 amino acids to 8,192 amino acids. * In the PEER benchmark test, Prot42 performs well in 14 tasks such as protein function prediction, subcellular localization, and interaction modeling. * Unlike AlphaProteo, which relies on 3D structure, Prot42 only needs the target protein sequence to generate binders.
Paper address:
More AI frontier papers:
https://go.hyper.ai/UuE1o
The open source project "awesome-ai4s" brings together more than 100 AI4S paper interpretations and provides massive data sets and tools:
https://github.com/hyperai/awesome-ai4s
Datasets: 3 large datasets support model development-training
In this study, several key data sets were used to train and evaluate the performance of its model. These data sets not only cover a wide range of protein sequence information, but also involve protein-DNA interaction data, providing rich training materials for Prot42.
Protein-DNA Interface Database (PDIdb) 2010
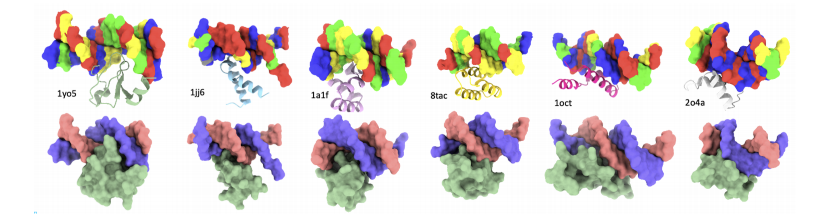
To design proteins that can bind to target DNA sequences, the researchers used the PDIdb 2010 dataset.This dataset contains 922 unique DNA–protein pairs and was used to train and evaluate the ability of Prot42 to generate proteins binding to specific DNA sequences.To evaluate the four DNA-protein models, the researchers extracted DNA fragments from various PDB structures, including 1TUP, 1BC8, 1YO5, 1L3L, 2O4A, 1OCT, 1A1F, and 1JJ6.
UniRef50 dataset
The pre-training dataset of the Prot42 model mainly comes from the UniRef50 database.The database contains 63.2 million amino acid sequences, covering a wide range of biological species and protein functions.These sequences are clustered, and sequences with similarity exceeding 50% are grouped together, thereby reducing data redundancy and improving training efficiency.
Before training Prot42, the research team preprocessed the UniRef50 dataset.They are labeled using a vocabulary of 20 standard amino acids.Use Xtoken to represent amino acid residues (X is used to mark uncommon or ambiguous amino acid residues).
In the data preprocessing stage,The research team processed the sequences with a maximum context length of 1,024 tokens and excluded sequences longer than this, ultimately obtaining a filtered dataset of 57.1 million sequences.The initial filling density is 27%. In order to improve data utilization and computational efficiency, the research team adopted a variable sequence length (VSL) filling strategy.We maximized the occupancy rate of tokens within a fixed context length, and finally reduced the dataset to 16.2 million padded sequences.The filling efficiency reaches 96%.
STRING database
The STRING database is a comprehensive protein-protein interaction database.It integrates experimental data, computational predictions, and text mining results to provide confidence scores for protein interactions. In order to train Prot42 to generate protein binders, the research team screened protein interaction pairs with confidence scores ≥ 90% from the STRING database to ensure the high reliability of the training data.Furthermore, sequence length was limited to 250 amino acids to focus on manageable single-domain binding proteins.After screening, the final dataset contains 74,066 protein-protein interaction pairs, a training set D(train)(pb) containing 59,252 samples and a validation set D(val)(pb) containing 14,814 samples.
Model architecture: 2 major variants derived from the autoregressive decoder architecture
Prot42 mentioned in this paper is a PLMs based on an autoregressive decoder architecture that generates amino acid sequences one by one and predicts the next amino acid using the previously generated amino acid. This architecture enables the model to capture long-distance dependencies in the sequence.It is able to learn rich representations directly from large unlabeled protein sequence databases, effectively bridging the gap between the huge number of known protein sequences and the relatively small proportion of protein sequences (<0.3%).At the same time, the model contains multiple Transformer layers, each of which contains a multi-head self-attention mechanism and a feedforward neural network to capture complex patterns in the sequence.
Its design is inspired by breakthroughs in natural language processing, especially the LLaMA model. Prot42 captures the evolutionary, structural, and functional information of proteins by pre-training on large-scale unlabeled protein sequences, thereby enabling the generation of high-affinity protein binders.
On this basis,The researchers pre-trained 2 model variants,That is, Prot42-B and Prot42-L.
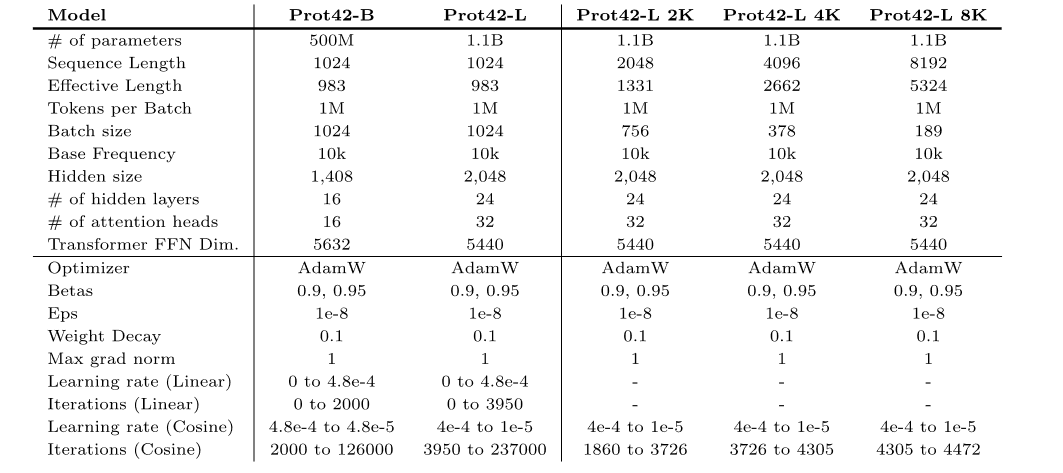
* Prot42-B:In the basic version, the model has 500 million parameters and supports a maximum sequence length of 1,024 amino acids.
* Prot42-L:The large version has 1.1 billion model parameters and also supports a maximum sequence length of 1,024 amino acids.The context length of Prot42-L was gradually extended from 1,024 amino acids to 8,192 amino acids.In this process, a gradually increasing context length and a constant batch size (1 million non-filled tokens) were used to ensure the stability and efficiency of the model when processing long sequences, significantly improving the model's ability to process long sequences and complex protein structures.Prot42-L also contains 24 hidden layers, each with 32 attention heads.The hidden layer dimension is 2,048.
Experimental conclusion: Great potential is shown in all 6 tasks
To evaluate the performance of the Prot42 model before validation on downstream tasks, the researchers used the standard measure of parametric complexity (PPL) for evaluating autoregressive language models, namely the performance of the Prot42 model at different context lengths.All models have relatively high perplexity at 1,024 tokens, but improve significantly to around 6.5 at 2,048 tokens.Results show that the base model and models fine-tuned for shorter contexts exhibit similar performance patterns across their respective maximum context lengths. The performance of the 8k context model is particularly striking - although its perplexity is slightly higher for medium-length sequences (2,048 – 4,096 tokens), it is able to handle sequences up to 8,192 tokens and achieves a minimum perplexity of 5.1 at the maximum length.After more than 4,096 tokens, the perplexity curve shows a downward trend.As shown in the figure below.
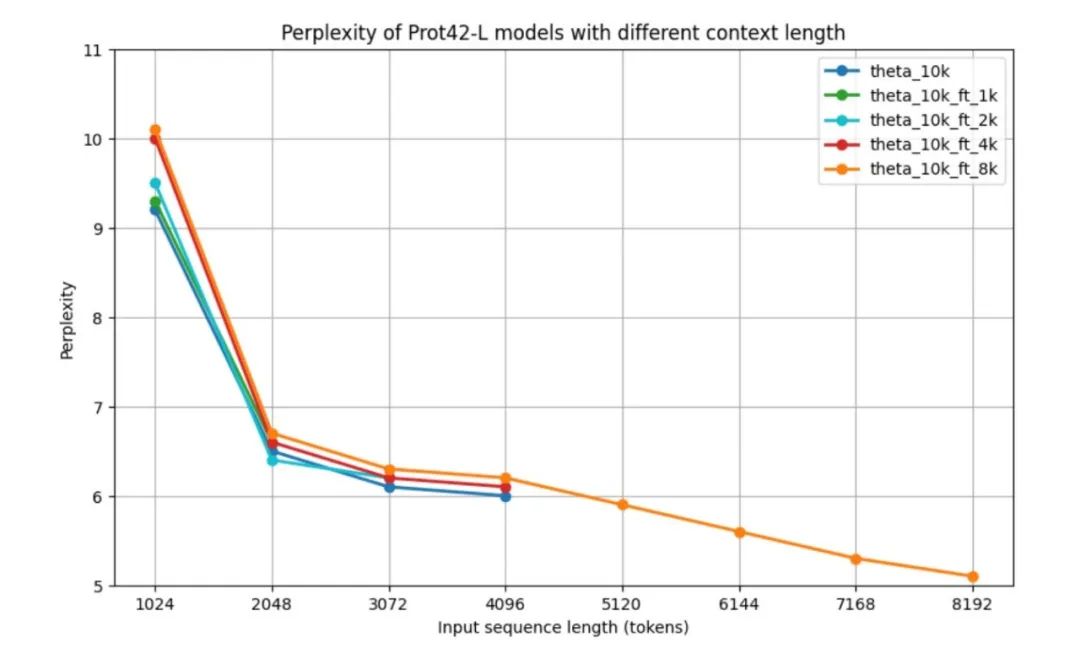
As the context length increases, the PPL of the model gradually decreases, indicating that the model's ability to process long sequences has been significantly improved.In particular, the 8K context model achieves the lowest PPL, indicating that it can effectively utilize the extended context window to capture long-range dependencies in protein sequences.The expanded context window is a major advance in the field of protein sequence modeling, enabling more accurate representation of complex proteins and protein–protein interactions, which is critical for generating effective protein binders.
Through a series of rigorous experimental evaluations,Prot42 has demonstrated excellent performance in multiple key tasks.It has been demonstrated to be effective in the generation of protein binders and the design of proteins binding to specific DNA sequences.
Protein function prediction
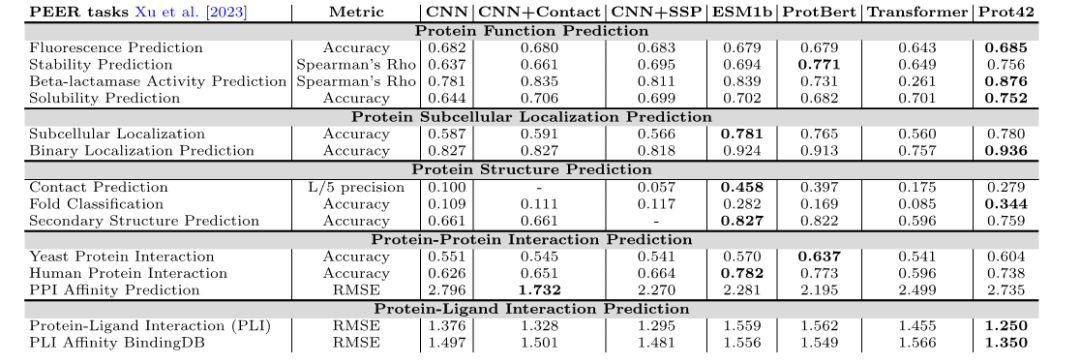
In the PEER benchmark test, the Prot42 model performed well in multiple protein function prediction tasks, including fluorescence prediction, stability prediction, β-lactamase activity prediction, and solubility prediction. Compared with existing models,Prot42 has achieved significant advantages in stability prediction, solubility prediction and β-lactamase activity prediction.This indicates its great potential in high-resolution protein engineering tasks.
Protein subcellular localization prediction
The researchers represented each protein sequence as a high-dimensional vector of size 32×2048, embedded the Prot42-L model in the entire protein sequence and performed calculations. In order to intuitively evaluate the differentiation of quality in embedding and compartments, the researchers applied t-distributed stochastic neighbor embedding (t-SNE) to reduce the dimensionality, making the visualization of protein groups clear.It has been verified that Prot42 performs well in the task of protein subcellular localization prediction, and its accuracy is comparable to that of existing advanced models.Through visual analysis, the research team further verified the effectiveness of the Prot42 model in capturing the subcellular localization characteristics of proteins.
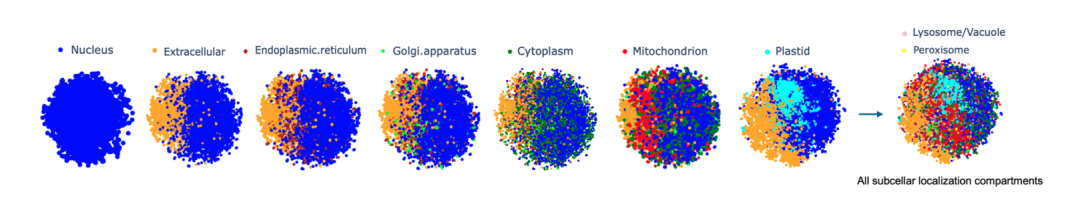
Protein structure prediction
In the protein structure prediction task,The Prot42 model achieved excellent results in contact prediction, folding classification, and secondary structure prediction.These results indicate that the Prot42 model is able to capture subtle differences in protein structure, providing strong support for complex biological interaction modeling and pharmaceutical applications.
Protein-protein interaction prediction
In the prediction tasks of protein-protein interaction and protein-ligand interaction, the Prot42 model showed high accuracy and reliability.The researchers used Chem42 to generate chemical embedding vectors and compared them with ChemBert., as another chemical representation model, even so, its performance indicators are still better than existing methods and close to the results achieved using Chem42. In particular, when using Chem42 to generate chemical embeddings, its prediction results are close to those of professional chemical models.This indicates that Prot42 has good extensibility in combining chemical information.Provides strong support for drug design.
Protein binder generation
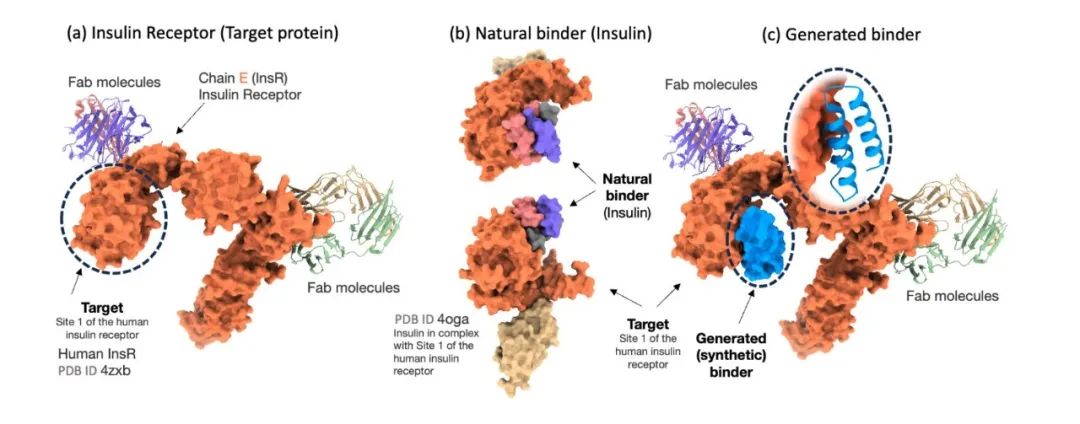
To rigorously evaluate the effectiveness of the Prot42 model in generating protein binders, the researchers compared the model with AlphaProteo, an advanced model designed specifically for protein binder prediction. The experimental results showed thatThe Prot42 model generated binders with strong predicted affinities on multiple therapeutically relevant targets.Especially on targets such as IL-7Rα, PD-L1, TrkA and VEGF-A,The Prot42 model performed significantly better than the AlphaProteo model.These results indicate that the Prot42 model has significant advantages in protein binder generation, as shown in the figure below.
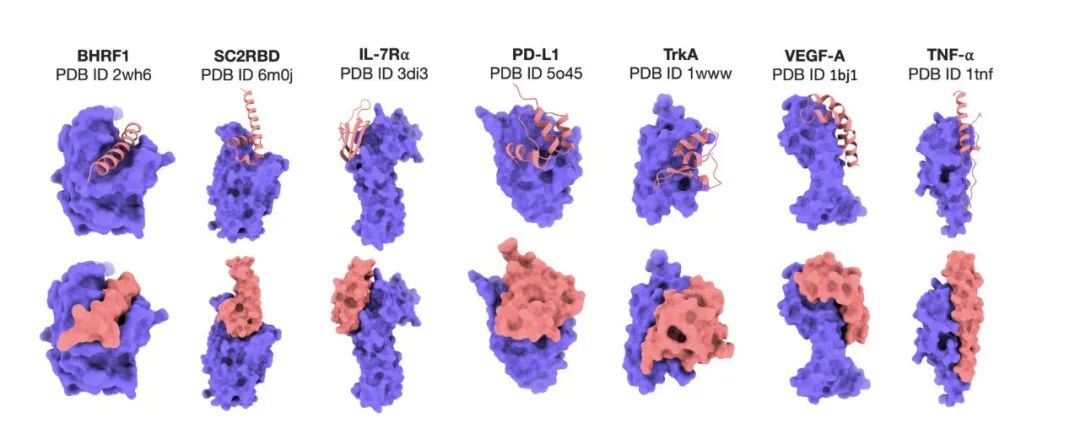
DNA sequence-specific binder generation
In the DNA sequence-specific binder generation experiment, Prot42 also achieved remarkable results. The experimental results showed thatBy combining a multimodal strategy of gene embedding and protein embedding, the Prot42 model is able to generate proteins that bind specifically to target DNA sequences and exhibit high affinity.The binding specificity evaluated by the DeepPBS model was high. These results indicate that the Prot42 model also has great potential in the generation of DNA sequence-specific binders, providing new tools for gene regulation and genome editing applications.
Breakthroughs and innovations in artificial intelligence in protein design
With the deep integration of biotechnology and artificial intelligence, the frontier field of protein design is undergoing revolutionary changes. As the core executor of life activities, the structural and functional analysis of proteins has always been a difficult point in scientific research, and the intervention of AI technology is accelerating the solution of this complex puzzle, opening up new paths for scenarios such as new drug development and enzyme engineering transformation.
In recent years, AI technology has made breakthroughs again, and new technologies centered on generative AI are pushing protein design into the "Genesis" stage.
Professor Xu Dong's team at the University of Missouri proposed the structure-aware protein language-aware model (S-PLM), which aligns protein sequence and 3D structure information into a unified latent space by introducing multi-view contrast learning.We use Swin Transformer to process the structural information predicted by AlphaFold and fuse it with the ESM2-based sequence embedding to create a structure-aware PLM.The article "S-PLM: Structure-Aware Protein Language Model via Contrastive Learning Between Sequence and Structure" was published in Advanced Science. S-PLM cleverly incorporates structural information into sequence representation by aligning protein sequences with their three-dimensional structures in a unified latent space. It also explores efficient fine-tuning strategies, enabling the model to achieve excellent performance in different protein prediction tasks, marking an important advancement in the field of protein structure and function prediction.
Paper address:
https://advanced.onlinelibrary.wiley.com/doi/10.1002/advs.202404212
In addition, the Tsinghua University research team and others proposed a unified protein language model xTrimoPGLM, which is a unified pre-training framework and basic model that can be expanded to 100 billion parameters and is designed for various protein-related tasks, including understanding and generation (or design). By utilizing the general language model (GLM) as the backbone of its bidirectional attention and autoregressive objectives, this model is different from previous encoder-only or causal decoding-only PLMs. This study explored the unified understanding and generation pre-training of ultra-large-scale PLMs, further revealed new possibilities for protein sequence design, and promoted the further development of a wider range of protein-related applications. It was published in the Nature sub-journal with the title "xTrimoPGLM: unified 100-billion-parameter pretrained transformer for deciphering the language of proteins".
Paper address:
https://www.nature.com/articles/s41592-025-02636-z
The breakthrough of Prot42 is not only a technical advancement, but also reflects the gradual maturity of the "data-driven + AI design" model in the field of life sciences. In the future, the research team plans to verify the binders generated by Prot42 through experiments and supplement computational evaluation with actual functional tests. This step will consolidate the utility of the model in practical applications and improve its predictive accuracy, bridging the gap between AI-driven sequence generation and experimental biotechnology.
References:
1.https://arxiv.org/abs/2504.04453
2.https://mp.weixin.qq.com/s/SDUsXpAc8mONsQPkUx4cvA
3.https://mp.weixin.qq.com/s/x7_Wnws35Qzf3J0kBapBGQ
4.https://mp.weixin.qq.com/s/SDUsXpAc8mONsQPkUx4cvA








