Command Palette
Search for a command to run...
Selected for ICML 2025, Tsinghua University/Renmin University of China/ByteDance Proposed the First cross-molecule Unified Generation Framework UniMoMo to Achieve multi-type Drug Molecule Design

The group led by Professor Liu Yang from Tsinghua University, the group led by Professor Huang Wenbing from the Gaoling School of Artificial Intelligence at Renmin University of China, and the ByteDance AI pharmaceutical team jointly proposed a unified generation framework across molecular species, UniMoMo.The framework unifies different types of molecules based on molecular fragments (blocks), compresses the full-atom conformation of each block using variational autoencoders, and performs geometric diffusion modeling in the compressed latent space, thereby achieving the design of different types of binding molecules (small molecules, peptides, antibodies) for the same target. UniMoMo has achieved leading performance in the evaluation of multiple molecular task benchmarks, demonstrating the great potential of cross-modal knowledge transfer and data sharing.

The related results were selected for ICML 2025 under the title "UniMoMo: Unified Generative Modeling of 3D Molecules for De Novo Binder Design".
Paper address:
Open source project address:
https://github.com/kxz18/UniMoMo
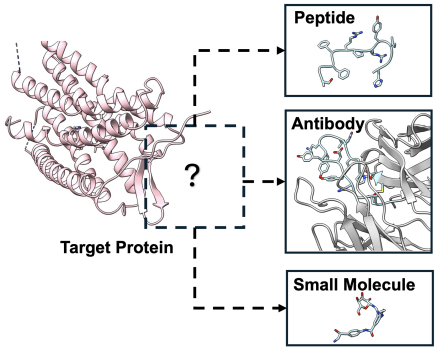
Why Unified Modeling?
Different molecular types have their own advantages and disadvantages in drug development, so it is often necessary to select the most appropriate molecular type in different disease scenarios. For example:
* Small molecules are small in size, easy to take orally, and have strong penetration, making them suitable for entering cells and acting on targets. They are widely used in chronic diseases and metabolic diseases;
* Peptide molecules have high targeting properties and can bind to large and flat areas on the surface of proteins. They are suitable for targeting "difficult-to-drugged" protein interaction sites and are often used in the treatment of cancer, inflammation, etc.
* Antibodies have extremely high selectivity and affinity, and can stably identify specific protein markers, making them particularly suitable for precise intervention scenarios such as immunotherapy.
Therefore, in the face of different disease mechanisms, target characteristics and drug requirements, the types of molecules suitable for use are different. Existing generation methods usually only model a certain type of molecule (such as small molecules, peptides or antibodies).It can neither meet diverse therapeutic needs nor utilize the commonalities between different molecules to improve model performance.
From an application perspective, unified modeling allows us to simultaneously explore multiple types of drug candidates for the same target, providing more options for different downstream scenarios.
From the perspective of machine learning, different types of molecules share similar binding rules (hydrogen bonds, π-π stacking, salt bridges, etc.) and geometric constraints (bond lengths, bond angles, etc.), and can learn from each other.Therefore, unified modeling should be able to improve the generalization and cross-transfer capabilities of the model by utilizing a larger data scale.
The Difficulty of Generative Unified Modeling
Although the idea of uniformly generating different types of molecules is exciting, there are still huge challenges in realizing such a framework, mainly in the choice of molecular representation and the design of the generation algorithm.
First, there are large differences in the structural representation of different molecular types: small molecules are composed of various functional groups, and their structures are highly diverse and nonlinear; peptides and antibodies are composed of amino acids connected in a linear sequence, and antibodies in particular have clear functional regions. An intuitive but ineffective approach is to model all molecules as atomic graphs.However, this approach ignores the natural hierarchical structure of molecules, such as key substructures such as benzene rings or standard amino acids, and leads to extremely high computational costs when dealing with systems with large binding surfaces such as antibodies.
On the contrary, if only common structural fragment vocabularies are used to construct fragment-level graphs (e.g., most protein design work only considers Cα coordinate),Ignoring atomic-level details will sacrifice the portability and accuracy of molecule generation.Because the essential laws of binding molecule design are the spatial interaction with the target and the geometric constraints within the molecule, these are physical laws defined at the atomic level and require precise all-atom information support.
Therefore, to build a truly effective and efficient unified molecular representation, two challenges must be solved simultaneously:It is necessary to retain the geometric details at the atomic level while abstracting the structural hierarchical priors.
Secondly, if structural fragments are introduced in the generation to preserve the hierarchical prior, it will bring core challenges to the generation algorithm:Traditional diffusion models usually rely on fixed-length, fixed-structure data representations.For example, a fixed number of point clouds or atoms. For structural prediction models such as AF3, since the 2D topology is given in advance, the number of atoms or 2D structure will not change during the diffusion process. For the task of molecular generation, the 2D topology and 3D structure need to be generated at the same time, and when the type of structural fragment changes during the denoising process, the corresponding number, type and arrangement of atoms will change accordingly. This breaks the assumptions of conventional diffusion models and places extremely high demands on modeling.
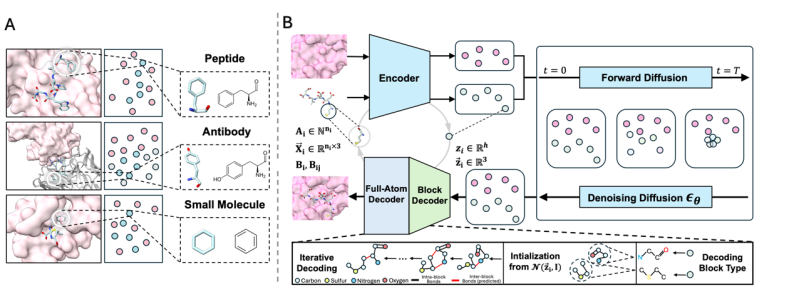
UniMoMo: A unified generative model
In order to solve the problem of large structural differences and high modeling difficulty of different molecular types, the article proposes a new framework - UniMoMo.It starts with two key designs, effectively taking into account the structural hierarchy and atomic-level precision:
* Unified representation:All molecule types are modeled in block form.
Whether it is a small molecule, peptide or antibody, UniMoMo represents its structure as a graph composed of molecular fragments (blocks). Each block can be a standard amino acid or a common small molecule fragment (such as a benzene ring, indole, etc.). In the implementation of the article, the recorded molecular fragments include all standard amino acids and small molecule fragments automatically identified by the principle subgraph mining algorithm. All non-natural amino acids can be treated as small molecules for word segmentation.This representation retains both the atomic-level details of the molecules and the hierarchical structure of different types of molecules themselves, making unified modeling possible.
* All-atom geometric implicit space diffusion model:Efficient generation on compressed representations.
In order to solve the problem of synchronous changes in the type and quantity of atoms caused by changes in block types during the generation process, and to improve generation efficiency and structural accuracy,The article designs an all-atomic iterative variational autoencoder (IterVAE).All atoms in each block are compressed into a "point" in the latent space, including a fixed-length latent space representation vector and the corresponding latent space coordinates.
The model then performs generative modeling in this compressed geometric latent space to generate latent representations of new molecules, which are finally decoded back to the full atomic structure.Since the data representation of the latent space is fixed-length (the number of blocks is pre-given) and continuous, it can be easily compatible with various existing generation algorithms.In the current attempts, the diffusion model has been able to produce relatively good results. This design allows the model to focus on the global layout between blocks during the generation process, while the detailed atomic-level structure is completed by the decoder, thus achieving the unity of high efficiency and atomic-level accuracy at the same time.
Unified Modeling Goes Beyond Single Domain Modeling
In order to verify the versatility and effectiveness of UniMoMo on different types of molecules, the authors conducted a systematic evaluation in multiple structure-based design tasks.It covers three representative types of binding molecules: small molecules, peptides and antibodies.By comparing with the most representative single-molecule type generation model in the corresponding field, the experiment aims to explore whether unified modeling has stronger geometric modeling capabilities and cross-modal generalization capabilities, especially in terms of key indicators such as spatial structure rationality and binding ability.
The results show thatUniMoMo, trained uniformly, has achieved comprehensive superiority in all molecular types.Not only does it excel in structural restoration accuracy, but it also achieves significant improvements in key geometric rationality and the quality of interaction with the target.
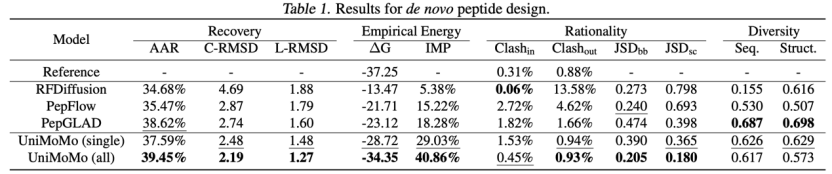
In the peptide generation task,UniMoMo significantly outperforms existing domain-specific models in multiple key indicators.Including RFDiffusion, PepFlow and PepGLAD, etc. Especially in terms of structural accuracy, UniMoMo achieved lower RMSD of complexes and monomers, indicating that the peptide structures it generated are closer to the real binding conformation.
UniMoMo can also generate structures with lower Rosetta binding energies.This reflects its stronger modeling ability for the geometric features of protein binding sites.In addition, UniMoMo also showed leading performance in geometric rationality indicators such as the consistency of dihedral angle distribution (JSD of backbone/sidechain torsions) and atomic-level spatial conflicts (clash rate) that measure the quality of peptide conformations. Moreover, UniMoMo (all) trained with all data consistently outperformed the model trained only with peptide data in various indicators.The ability of UniMoMo to learn and generalize across molecular species is demonstrated.
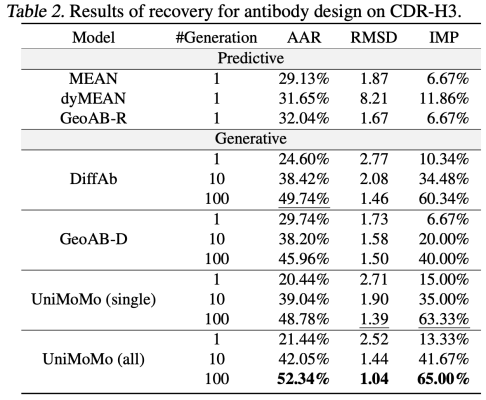
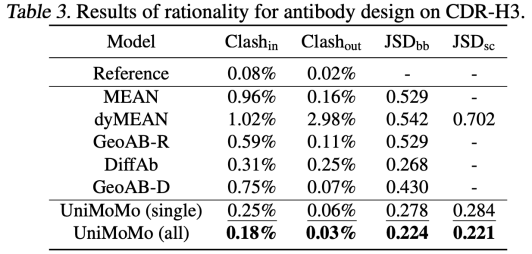
In the antibody design task, UniMoMo also shows strong performance. Compared with existing methods such as MEAN, dyMEAN and DiffAb,UniMoMo surpasses all other targets in terms of key indicators such as recall of naturally bound sequences and structures (AAR and RMSD) and binding energy improvement (IMP).Especially in the evaluation of multiple sampling generation, UniMoMo is able to generate antibody fragments close to the natural conformation with a higher probability, showing its good exploration ability in the antibody structure space.
Similarly, UniMoMo(all), which is jointly trained using data from different molecular types, outperforms the version trained only using antibody data in all indicators.This shows that unified modeling does help the model learn more universal and transferable spatial laws of molecular structures.This result highlights the commonalities in structural modeling between different molecular types and verifies the significant value of cross-domain data fusion in improving generation quality.
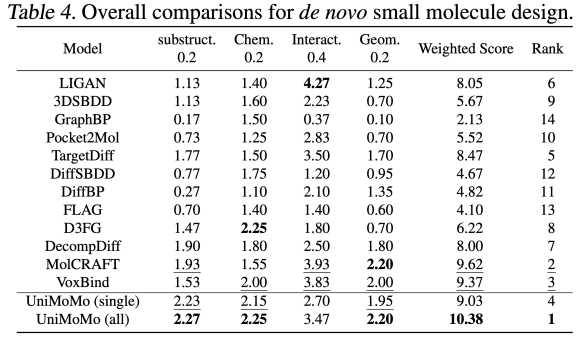
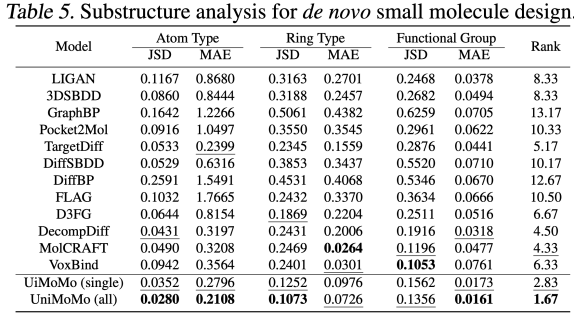
In the small molecule generation task, UniMoMo also demonstrated superior performance. Through evaluation on the CrossDocked2020 dataset,The authors found that UniMoMo surpassed existing mainstream methods in comprehensive evaluation based on CBGBench.
Specifically, UniMoMo has achieved higher comprehensive scores in terms of substructure distribution (atomic types, functional groups, etc.), chemical property rationality (QED, LogP, SA, etc.), geometric structure quality (bond length/angle distribution and atomic conflict rate, etc.), and interaction score (Vina docking) (please refer to the original text for complete experimental results). More importantly, compared with the single-domain version trained only on small molecule data, UniMoMo (all) trained across molecule types has significant improvements in all evaluation dimensions. This shows thatEven in the small molecule scenario with the most flexible molecular structure and the most diverse types, the unified model can still transfer geometric laws and interaction patterns from other molecular types, thereby improving the rationality of the monomer conformation and relative pocket space layout of the small molecule.This phenomenon once again verifies the core concept of UniMoMo: the geometric constraints and binding mechanisms between different molecules have shareable patterns, and unified modeling can effectively stimulate this potential.
Combining the experimental results of the three types of tasks, UniMoMo shows highly consistent advantages: the unified model trained with cross-molecule data outperforms the existing single-domain generative model in each task, and has a significant improvement over UniMoMo trained with only single-domain data. This phenomenon shows that the underlying physical and chemical constraints and spatial geometric laws of seemingly different tasks in molecular design are actually highly common.UniMoMo's unified modeling strategy captures and amplifies this commonality, thereby achieving cross-task transfer and complementary enhancement.These results not only verify the effectiveness of UniMoMo, but also provide strong empirical support for building a more powerful unified molecular generation system in the future.
GPCR Case Studies
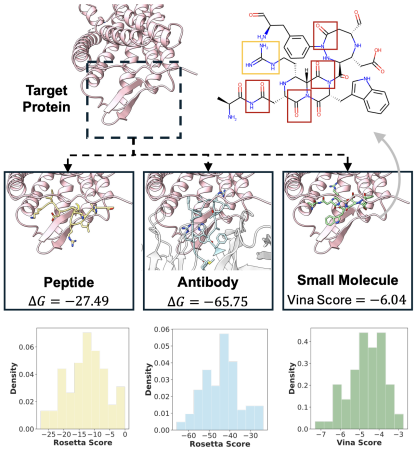
As a case study, the authors selected one of the most important drug targets in humans, the G protein-coupled receptor (GPCR), to evaluate UniMoMo's ability to generate different types of molecules (peptides, antibodies, small molecules) at the same binding site. The peptides, antibodies and small molecules generated by UniMoMo all show good distribution under the force fields commonly used for binding energy evaluation (Rosetta ΔG, Vina score).What is even more surprising is that the generated small molecule structure also spontaneously simulates functional groups similar to natural amino acid side chains, which are used to build hydrogen bonds and form key interactions with the target. In addition, small molecules also borrow local geometric configurations from peptides and antibodies, such as amide connections on the molecular skeleton, so that they can effectively fill binding pockets that are originally more suitable for large molecules. This case vividly demonstrates UniMoMo's ability to cross-modal reference and automatically adapt to binding pockets in actual tasks, and reflects its potential to deeply understand the interaction between targets and molecules and the internal geometric constraints of molecules at the three-dimensional structural level.
Future exploration
Although UniMoMo has demonstrated strong unified generation capabilities in multiple molecular types and tasks, the authors also pointed out that there are still many future possibilities worth exploring in this direction.
The current work mainly focuses on the modeling of natural amino acids and common molecular fragments, which can be further expanded to non-natural amino acids, post-modified peptides/antibodies, cyclic molecules and other more complex drug forms, thereby covering a wider range of candidate molecular spaces. The concept of unified modeling also provides an opportunity for the study of the controllability and interpretability of the model, and is expected to further promote the development of generative models into more reliable and practical molecular design platforms. In short, the introduction of UniMoMo not only provides a general and powerful generative framework for molecular design tasks, but also opens up a new direction full of potential for AI-driven drug discovery.








