Command Palette
Search for a command to run...
MIT and Harvard Jointly Proposed PUPS to Achieve single-cell Protein Localization by Integrating Protein Language Model and Image Inpainting Model
Subcellular localization of a protein refers to the specific location of a protein in the cell structure.This is crucial for proteins to perform their biological functions. For example, if you imagine a cell as a huge enterprise, where the nucleus, mitochondria, cell membrane, etc. correspond to different departments such as the president's office, power generation department, and gate guard, then only when the corresponding protein enters the correct "department" can it work normally, otherwise it will cause certain diseases, such as cancer and Alzheimer's disease. Therefore, precise positioning of protein subcellular can be said to be one of the core tasks of life science.
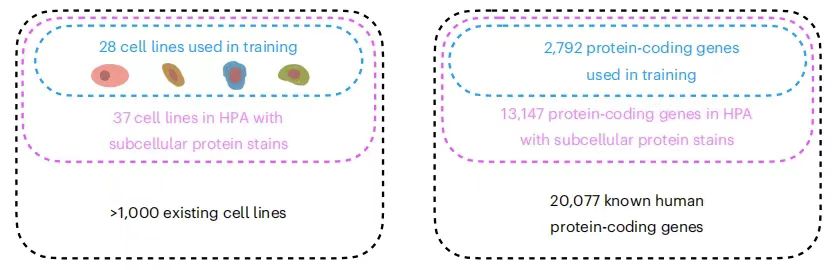
Although the research community has performed spatial localization analysis on thousands of proteins in different cell lines, the number of protein and cell line combinations that have been measured so far is only the tip of the iceberg.The Human Protein Atlas (HPA) provides the subcellular localization of proteins encoded by 13,147 genes (accounting for 65% of known human protein-coding genes).However, the entire dataset contains 37 cell lines, and each protein can only be measured in three of them at most. At the same time, mainstream experimental methods are difficult to detect all the protein numbers in the same cell at the same time, which seriously hinders the comprehensive analysis of complex protein networks and increases the complexity of the experiment and the risk of error.
In addition, protein localization is not static and varies not only between cell lines but also between individual cells in the same cell line, and the protein and cell line pairs recorded in existing data maps only reflect the results under specific conditions.Even existing results are difficult to apply directly, and further exploration of protein localization is needed based on environmental changes.
In order to solve the contradiction between the limitations of protein subcellular localization technology and the complexity of biological systems, machine learning is expected to be successful. Models based on protein sequences and cell images have been successfully applied, and although they have performed well in some aspects, their shortcomings are also very prominent - the former ignores the specific localization differences of cell types, and the latter lacks the generalization ability to promote the study of unknown proteins.
In view of this,A research team from the Massachusetts Institute of Technology and Harvard University proposed a prediction framework for the subcellular localization of unknown proteins by combining protein sequences and cell images, named Predictions of Unseen Proteins' Subcellular localization (PUPS). PUPS innovatively combines protein language models and image restoration models to predict protein localization, which enables it to combine the generalization ability of unknown protein prediction and cell type-specific prediction that captures cell variability. Experiments have shown that the framework can accurately predict the localization of proteins in new experiments outside the training dataset, with excellent generalization ability and high accuracy, and outstanding application potential.
The research results, titled "Prediction of protein subcellular localization in single cells," have been published in Nature Methods.
Research highlights:
* The proposed study innovatively combines protein language models and image rendering models, using protein sequences and cell images to predict protein localization, making up for the shortcomings of previous computational models
* PUPS is able to generalize to unknown proteins and cell lines, thereby assessing the variability of protein localization between cell lines and between individual cells within a cell line, and identifying biological processes associated with proteins with variable localization
* In new experiments outside the training dataset, PUPS also demonstrated its highly accurate prediction ability, with outstanding application potential and medical value

Paper address:
Datasets: Building trustworthy models with the most comprehensive data possible
The training data set of PUPS comes from the Human Protein Atlas (HPA).The research team aggregated the 16th version of HPA data into the 22nd version to collect as much protein data as possible and ensure the comprehensiveness of the experimental analysis. As shown in the figure below:
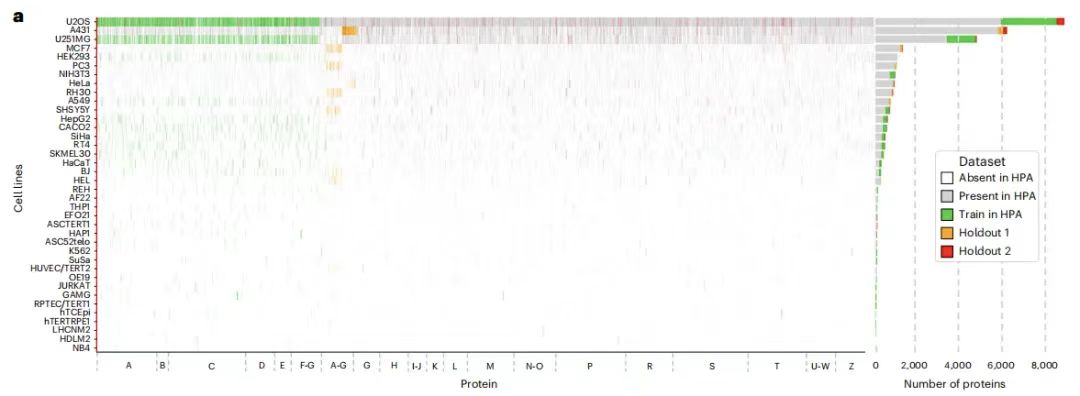
Specifically, the training dataset contains 340,553 cell numbers and 8,086 protein variants, corresponding to 2,801 genes in 37 HPA cell lines, whose names begin with the letters AG. In addition, the training dataset also includes 10 additional genes, including IHO1, IMPAD1, INKA1, ISPD, ITPRID1, KIAA1211L, KIAA1324, LRATD1, SCYL3, and TSPAN6.
The holdout dataset is divided into two parts:One part is reserved data set 1,It contains 36,552 cells, with 9,472 protein variants corresponding to 3,312 genes (including 2,801 in the training set), and their names also start with AG, but they come from different cell lines and have no overlap with the training set. At the same time, the reserved dataset 1 is further split into two parts, used as the evaluation set and the test set, containing 11,050 and 25,502 cells respectively;The retained dataset 2 contains 24,007 cells, corresponding to 515 genes.Its name starts with all letters of the alphabet, covering AZ. There are 556 protein variants in total, which come from new gene families that do not appear in the training set and the reserved dataset1 and can be used to test the generalization ability of the model.
It should be noted that the BJ cell line images were retained in both the training set and the holdout dataset 1.
Before the experiment, the research team preprocessed the images in HPA, which simply included the following five steps:
* Step 1,Each image was downsampled 4 times, and the final resolution was reduced to 0.32 μm/pixel in order to reduce the amount of calculation and remove high-frequency noise;
* Step 2,Gaussian blur (σ=5) and Otsu thresholding were combined to separate the approximate area of the cell nucleus from the complex background;
* Step 3, use the remove_small_holes function to remove holes with an area smaller than 300 pixels, then binarize the image and remove noise areas smaller than 100 pixels;
* Step 4,The centroid of each cell nucleus was calculated, and a 128 × 128 pixel area was cropped out with the centroid as the ROI of a single cell;
* Step 5,Through intensity normalization and noise filtering, standardized data distribution is achieved and inter-channel interference is reduced.
Model architecture: Combining protein sequence and image representation to predict protein subcellular localization
The PUPS model mainly consists of two parts:One is used to learn sequence representation from the amino acid sequence of the protein; the other is used to learn image representation from the iconic staining of the target cell.Then, the protein sequence representation and image representation are combined to predict the subcellular localization of the protein in the target cell. The former enables the model to be generalized to unknown protein predictions, and the latter enables the model to capture the variability at the single-cell level and realize cell type-specific localization predictions. As shown in the figure below:
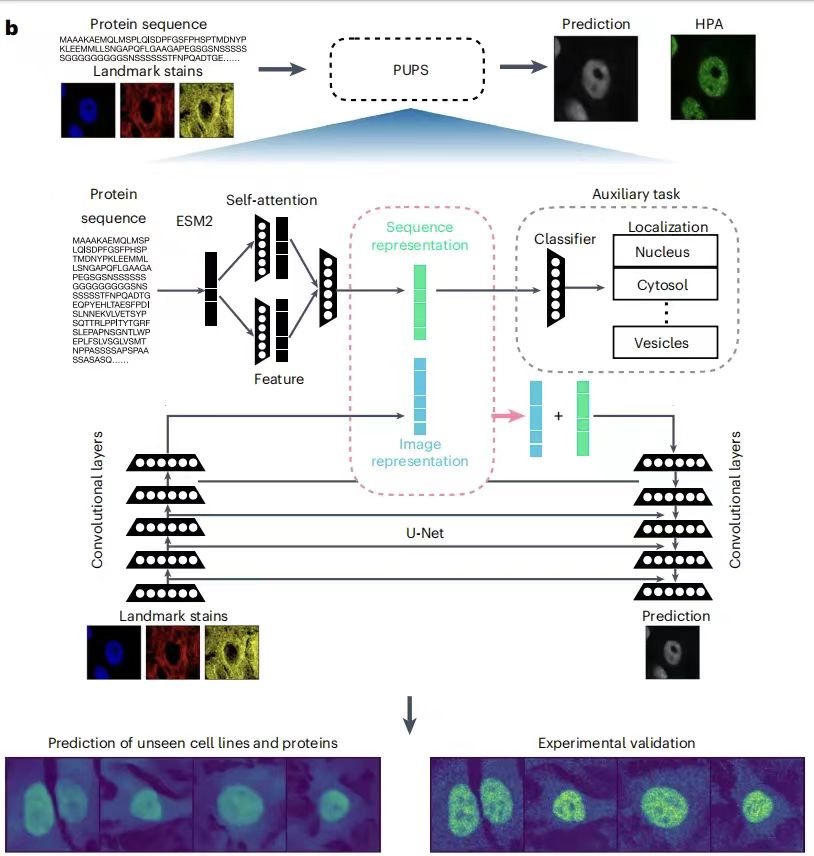
In simple terms,PUPS uses the pre-trained ESM-2 (Evolutionary Scale Modeling) protein language model to extract protein sequence features, and uses a convolutional neural network to learn the iconic staining image features of cells. Finally, the two parts of information are combined to predict the localization of proteins in target cells.It should be noted that all parts of the model are trained simultaneously, which helps to reduce the classification loss of the pre-task and the difference between the predicted protein images and the experimentally measured protein images in HPA. All parameters are optimized using the Adam optimizer with a learning rate of 1e-4.
Protein Language Model
PUPS learns sequence representations by using a language model, a self-attention layer, and an auxiliary pre-training task, and then classifies protein localization based on the learned sequence representations.
Specifically, the research team obtained the initial representation of specific protein variants by inputting the N-terminal 2,000 amino acid sequence into the pre-trained ESM-2 model, thereby generating a 1,280-dimensional vector for each amino acid residue, and zero padding was used for variants with less than 2,000 residues. This sequence length truncation is to avoid biased predictions for a small number of proteins with sequence lengths of up to tens of thousands of residues. As shown in the figure below:
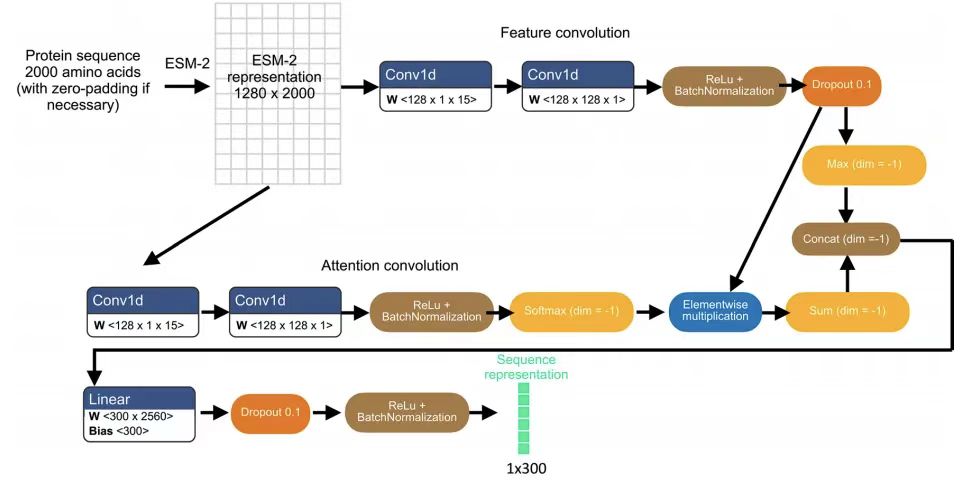
To adapt ESM-2 characterization for protein localization prediction,The team subsequently adopted a light attention layer of separable convolutions.The final 300-dimensional sequence representation is obtained by applying the ESM-2 representation. This protein sequence representation is used both for the auxiliary pre-task of predicting localization labels and for protein image prediction in combination with the image representation. The pre-task inputs the protein sequence representation into a fully connected neural network layer to input a 29-dimensional vector representing the probability distribution of 29 subcellular compartment localization labels, and then compares the pre-task output with the HPA annotated protein compartments using a binary cross entropy loss with sigmoid activation.
Image rendering model
The image input of each cell contains three iconic staining image channels: cell nucleus, microtubules and endoplasmic reticulum staining.Its dimensions are 3 x 128 x 128 and are centered at the nucleus centroid.
Image encoding is achieved through 5 separable convolutional layers.The final dimension is 16 x 16 x 512. Each convolutional layer is followed by leakyRelu activation, batch normalization, and 2D max pooling layers. The protein sequence representation is concatenated to all spatial dimensions of the cell image representation and then fed into a U-Net image decoder to learn different weights for each input channel. In addition, the spatial dimension weighting mechanism in the model allows each spatial dimension of the image representation to be combined with the sequence representation with different weights.
The decoder consists of 5 separable convolutional layers.The output of the image is 1 x 128 x 128, which is the protein image prediction of the corresponding cell. Then, skip connections similar to the image segmentation U-Net are added between the encoding layer that generates the image representation of the marker staining and the decoding layer that generates the protein image prediction at the same depth. The mean squared error loss function is used to train the model to minimize the difference between the predicted protein image and the experimentally measured protein image.
Experimental results: Achieving precise subcellular localization of proteins at the single-cell level
In order to verify the feasibility and effectiveness of the model, the research team proposed a number of experiments for verification. PUPS showed good performance in multiple tasks, highlighting its advantages in multi-model fusion.
Predicting variability in protein localization between cell lines
To evaluate the performance of PUPS in quantifying protein localization variability among cell lines,The research team calculated the protein nuclear ratio to quantify the localization variability and found that the predicted values were highly correlated with the real data.The Pearson correlation coefficient of Holdout 1 is 0.794, and the Pearson correlation coefficient of Holdout 2 is 0.878. As shown in the following figure:
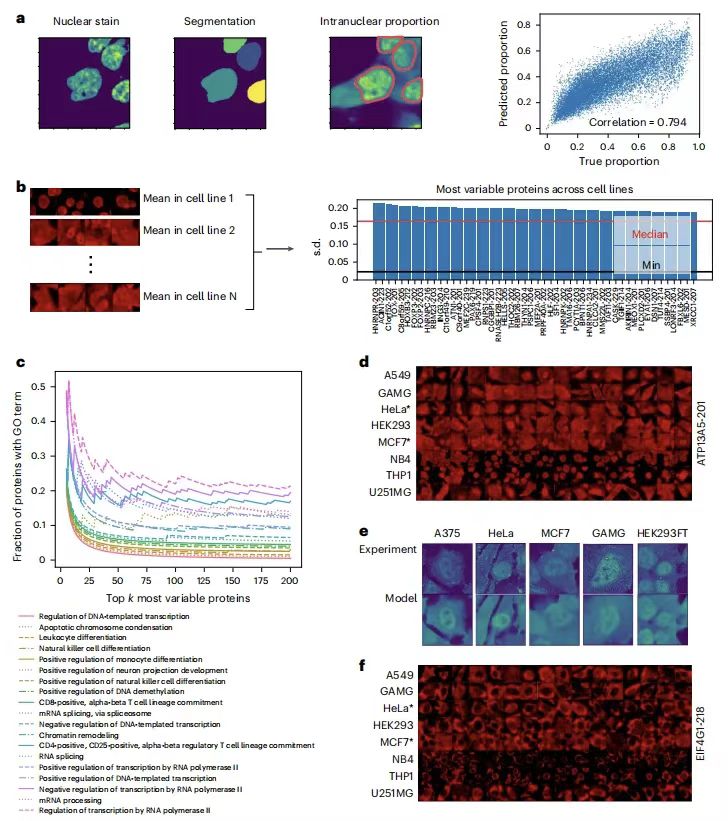
Subsequent further analysis showed that the proteins with the greatest localization variation between cell lines were associated with biological processes such as transcription, cell differentiation, and chromatin regulation, such as ATP13A5, which confirmed the accuracy of the model predictions.The model captures differences in cell morphology through signature staining and can infer cell line specificity of protein localization without cell line labels, providing a new method for studying the cell-specific regulation of protein function.
Predicting differences in protein localization between single cells
To evaluate the ability of PUPS to predict the variability of protein localization between single cells in the same cell line, the research team calculated the variance of the nuclear ratio of proteins in all single cells in each cell line.The results showed that the single-cell variability prediction ranking for each protein-cell line pair was highly consistent with the real data.For example, the overlap rate of the first 500 high-variant pairs in Holdout 2 exceeded 60%, and the predicted intranuclear ratio distribution was consistent with the actual results, eliminating the influence of prediction errors.
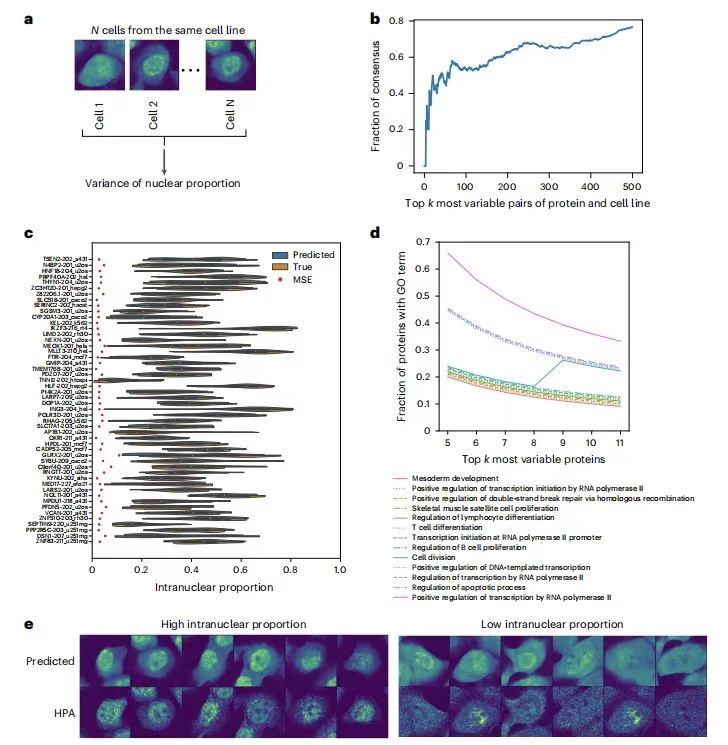
In addition, Gene ontology (GO) analysis showed that highly variable proteins were related to processes such as cell division, transcription, double-strand break repair, and apoptosis.The model captures morphological features through cell-marking staining images, indicating that single-cell variability is not only random but also related to cell morphological features.Provides a new perspective for explaining the mechanism of single-cell heterogeneity.
Validation of PUPS in new experiments outside the training data
In order to verify the ubiquitination ability of PUPS to predict protein localization in a new experimental environment, the research team selected 9 proteins for verification in 5 cell lines. As shown in the figure below:
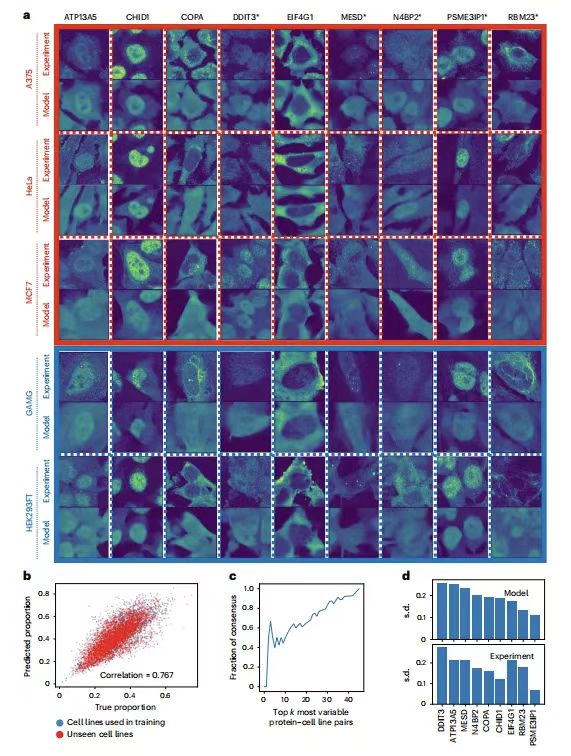
ATP13A5, CHID1, COPA, MESD and RBM23 are the proteins with the greatest variation among cell lines, and they all have different GO terms; DDIT3 and N4BP2 are the proteins with the greatest variation in individual cells within a cell line; EIF4G1 and PSME3IP1 are the proteins with the least variation among cell lines, the former is expected to be mainly located outside the nucleus, and the latter is expected to be mainly located inside the nucleus. Among the five cell lines, except A375, the other HeLa, MCF7, GAMG and HEK293FT are all included in HPA.
The results show thatThe protein images predicted by PUPS are visually similar to those measured experimentally.The nuclear protein ratio of each single cell calculated using the predicted protein image is closely related to the ratio calculated from the experimental measurement image, with a Pearson correlation coefficient of 0.767. This indicates thatPUPS can be used to quantitatively predict the localization of proteins that have not been previously measured experimentally or used in training atlases.
PUPS learns meaningful protein and cell representations
Experiments demonstrate that PUPS’s ability to predict protein localization in unknown proteins and cell lines comes from learning meaningful representations of protein sequences and cell landmark images.
The research team mapped protein sequence representations of 40,622 protein forms corresponding to 12,614 genes. Proteins with similar localizations tend to have similar sequence representations. To further demonstrate that the model can identify meaningful protein sequence patterns and predict localization, the research team used the Positional Shapley method to calculate the importance of each amino acid residue in a specific protein for predicting the label of each cell compartment. For example, the predicted variability of N4BP2 nuclear localization was successfully explained, which is consistent with reports that CUE domains may change subcellular localization through ubiquitin binding.
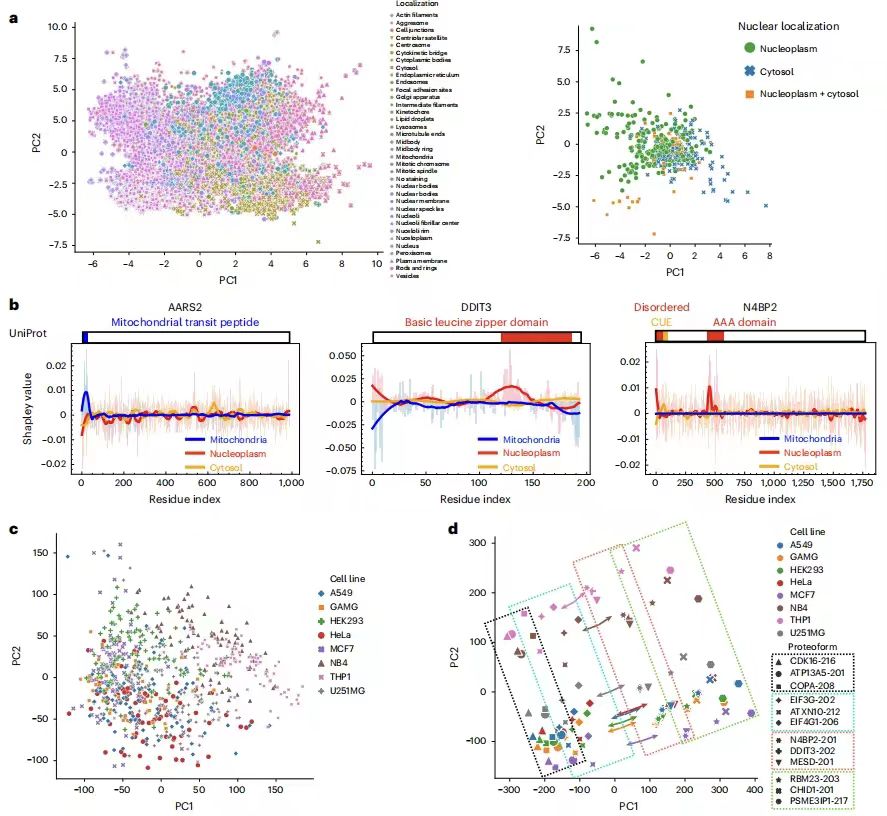
In addition to identifying meaningful protein sequence motifs,The research team further demonstrated that PUPS learns meaningful representations of single cells from cell-signature staining.It visualizes the single-cell image representations learned from landmark staining and finds that single cells of the same cell line have similar image representations even if the cell line label is not input into the model. The joint representation of protein and cell landmark images preserves the separation between cell lines and proteins, while the order of different proteins within each cell line is similar between different cell lines. Given the centroid of each cell line in the joint representation space, the vector from the centroid to a specific protein is mostly parallel in all cell lines, that is, given a sequence representation, predicting the image of a specific protein needs to move in the same direction in the representation space, regardless of the cell line.This explains the ability of PUPS to generalize to unknown proteins and cell lines by learning meaningful representations of protein and cell images.
also,PUPS can also predict the effects of disease-causing mutations on protein localization.For example, mutation studies on the nuclear-encoded mitochondrial proteins SDHD and ETHE1 showed that SDHD mutations lead to an increase in its nuclear localization ratio, which is consistent with the mechanism of nuclear genome instability in the disease; ETHE1 mutations show an increase in cytoplasmic localization ratio, which is associated with known nuclear-cytoplasmic shuttling abnormalities. These results indicate that PUPS can provide new clues for the study of disease mechanisms by analyzing the effects of sequence variation on localization.
New solution for protein subcellular localization prediction
As mentioned above, protein subcellular localization prediction is of great significance in bioinformatics and biological research. PUPS provides a way to integrate multimodal information, which makes a strong contribution to the research in this field. At the same time, after decades of development, the research in this field has already achieved great results.
A team from University College Dublin in Ireland published a study in the Computational and Structural Biotechology Journal.It introduces a variety of computational methods for protein subcellular localization prediction, including sequence-based, annotation-based, hybrid and meta-prediction-based methods. The article also classifies and introduces subcellular localization prediction tools by eukaryotes, prokaryotes, viruses and multiple categories.Eukaryotic prediction tools include mLASSO-Hum and DeepPSL, and prokaryotic prediction tools include PRED-LIPO. By designing machine learning and deep learning classification diagrams covering 7 major areas and 28 subcategories, this study provides a classification method for single-category and multi-category prediction tools, making it easier for users to find methods and prediction tools. The paper was published as "Protein subcellular localization prediction tools."
* Paper address:
https://www.sciencedirect.com/science/article/pii/S2001037024001156
On April 12, Yang Li's research group at the Institute of Biomedical Sciences, Fudan University, and Dong Nanqing's research group at the Shanghai Artificial Intelligence Laboratory, collaborated to publish an online research paper titled "Deep Generative Model for Protein Subcellular Localization" in the journal Briefings in Bioinformatics.The study also developed a generative deep learning model deepGPS with multimodal processing capabilities based on the ESM2 protein language model and U-Net framework.
According to reports, deepGPS can receive protein sequences and cell nucleus images as input and generate text labels and distribution images of protein localization. It is a new "text-to-image" multimodal model that supports the prediction of protein subcellular localization.
* Paper address:
https://doi.org/10.1093/bib/bbaf152
As the integration of artificial intelligence and biological research accelerates, related innovative experiments are constantly emerging, gradually breaking the drawbacks of traditional methods, achieving "the best of both worlds" or even "perfect" performance, thus promoting the rapid development of bioinformatics.








