Command Palette
Search for a command to run...
For the First Time, a Team From Columbia University Proposed PXRDnet to Analyze Nanocrystals end-to-end and Successfully Analyzed 200 Complex Simulated nanocrystals.

The discovery and application of X-ray diffraction (XRD) is an important milestone in the development of crystallography, because this technology enables people to gain a deep understanding of the microstructure of crystals, which in turn drives the progress of materials science and human civilization as a whole. However, when traditional methods encounter powdered nanocrystals composed of tiny particles, the ideal result does not occur.
Due to the limited size of nanocrystals (usually less than 1000 Å),The Bragg peak in its X-ray diffraction pattern shows obvious broadening.This leads to serious substantial degradation of structural information, which poses a huge challenge to accurately analyzing its crystal structure. In addition, the difficulty in obtaining pure single crystal samples in actual situations further increases the difficulty of structural analysis. Nanocrystal structure analysis has also become a "century-old problem" that has plagued the materials science community for a century.
To address this issue, researchers from Columbia University and Stanford University proposed a generative artificial intelligence structure analysis method PXRDnet based on a diffusion model.The model uses 45,229 known crystal structures as training data and introduces statistical prior knowledge.Even with only the chemical formula and the information-scarce limited-size broadened powder diffraction pattern as conditions, PXRDnet was able to successfully resolve 200 simulated nanocrystals of varying symmetry and complexity.Structures from all seven crystal systems are included, down to 10 Å in size.Experimental results show that the model can successfully and verifiably identify 4 out of 5 structural candidates, with an average error of only 7% after measurement by the Rietveld refinement r factor.
The related research was published in Nature Materials under the title "Ab initio structure solutions from nanocrystalline powder diffraction data via diffusion models".
Research highlights:
* This achievement has solved the long-standing problem of nanocrystal structure analysis in the materials science community and provided an efficient artificial intelligence analysis tool, which is expected to promote innovative applications in nanotechnology, biomedicine, energy storage, electronic devices and other fields
* This method significantly breaks through the applicability boundaries of traditional methods and obtains candidate solutions close to the real structure in many cases
* The study proposed the MP-20-PXRD benchmark data set (including stable materials with less than 20 atoms in the Materials Project and their simulated diffraction data), and made the code and data set public, providing a unified standard for subsequent research

Paper address:
https://go.hyper.ai/r1K6b
Materials Project online materials database:
https://go.hyper.ai/2gCe9
Dataset: Proposed MP-20-PXRD benchmark dataset
To obtain an effective model, the researchers provided a benchmark dataset called MP-20-PXRD for end-to-end training of PXRDnet.
Specifically, the researchers used the MP-20 dataset from the Materials Project.The dataset consists of materials sampled from the Materials Project database with a maximum of 20 atoms in the unit cell.The researchers then used the pymatgen package to simulate the powder diffraction patterns of all structures in MP-20.
Materials Project online materials database:
https://go.hyper.ai/2gCe9
The simulations used Cu Kα radiation with a Q range of 0-8.1568 Å⁻¹.
The MP-20-PXRD dataset contains 45,229 materials.The ratio of 90%, 7.5%, and 2.5% was used for training, validation, and testing. It is worth mentioning that the MP-20-PXRD dataset has been open sourced, and researchers hope to use it to inspire "latecomers" to further explore new solutions for nanocrystal structure analysis.
Model architecture: Based on CDVAE, introducing PXRD regressor
The PXRDnet model is designed based on the CDVAE architecture.It mainly consists of three main branches, namely, atomic denoising branch, variational autoencoder (VAE) branch and PXRD regressor.They are connected by a shared Gaussian latent code. This method enables PXRDnet to accurately generate qualified material structure candidates given a PXRD pattern and chemical formula, providing new insights into nanomaterial structure analysis.
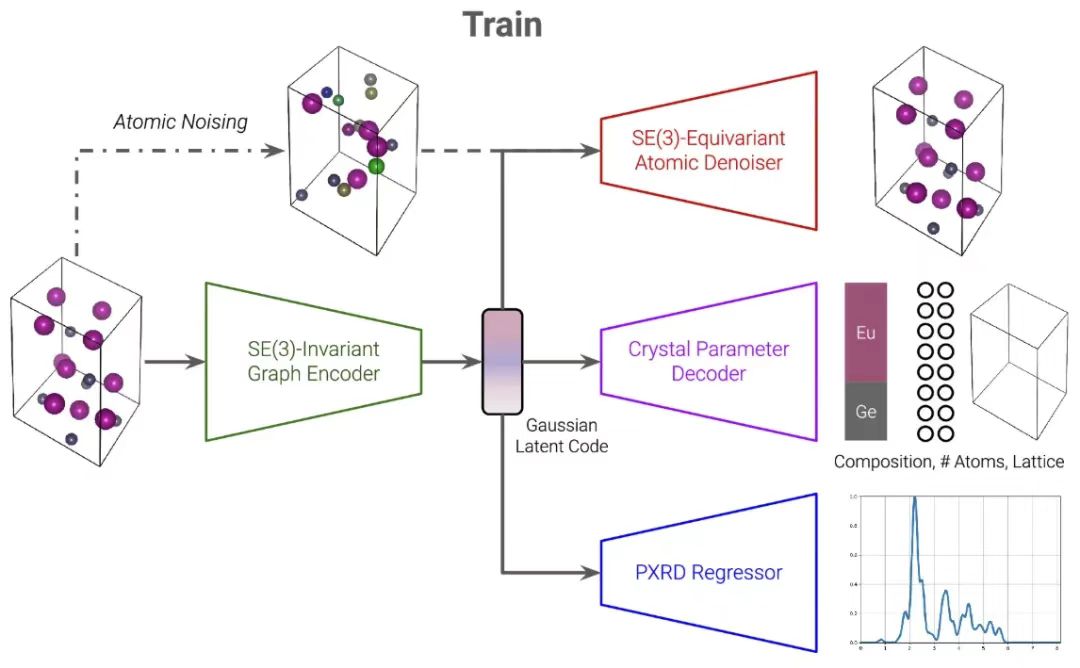
Skeleton development based on CDVAE
When introducing PXRDnet, we have to mention the CDVAE model, which is the basis for the creation of the former.CDVAE is a material structure generation model.It is inspired by variational autoencoders and denoising diffusion networks and is a generative model that learns to decompress data from noise.
To understand the decomposition of VAE and diffusion components, the researchers realized that the unit cell of a material can be represented by four components: chemical composition, number of atoms, lattice parameters, and atomic coordinates.
The first branch of CDVAE uses VAE to process the first three components.The encoder is DimeNet, a SE(3)-invariant Graph Neural Network that maps the graph representation of the material to a latent representation z. The graph representation is modified into a directed multigraph to reflect the inherent periodicity of the material. The researchers then regularize the latent representation z to a multivariate Gaussian distribution using the Kullback-Leibler divergence loss, and then decode the chemical composition, atomic number, and lattice parameters from z.
Each prediction is generated by a separate crystal-parameterized multilayer perceptron (MLP) that receives the latent code z.z will be used as the material representation in all other branches of the subsequent model.
The second branch of CDVAE utilizes denoising diffusion to process components through a noise-conditioned score network.It assumes that the number of component atoms and lattice parameters are fixed. The forward process perturbs the atomic coordinates and atomic species with multivariate Gaussian noise. The reverse process is parameterized by GemNet, an SE(3)-equivariant graph neural network. The process is conditioned on the above latent code z, which is the basis for its normal operation.
It is worth mentioning thatThe reverse process is essentially to predict how to denoise the perturbed atomic coordinates and species via Langevin dynamics.They are moved to their real positions and restored to their real species. Also the output graph representation is a directed multigraph, compatible with the periodicity of the material.
In the generation phase, CDVAE first samples a latent code z ≈ N (0, I) from a multivariate Gaussian distribution.The crystal parameter multilayer perceptron is used to decode it and obtain the component chemical composition, atomic number and lattice parameters, which can be used to initialize a unit cell, where the atomic positions are also randomly selected from N (0, I). The atomic positions and types are then optimized through the Langevin dynamics SE (3) equivariant image denoising process. During the entire denoising process, the lattice parameters and atomic number remain unchanged, and the resulting material is finally obtained.
Specially designed PXRD regressor
In addition, in this study, the powder X-ray diffraction (PXRD) pattern was set as the desired property to be predicted, so the researchers designed a PXRD regressor Fψ, which transforms the potential material representation z∈R256 Mapped to a vector y∈R512, i.e., the estimated Q-space characterization of the material's PXRD pattern.
The PXRD regressor is parameterized by a DenseNet-inspired architecture.This architecture extends the traditional convolutional neural network.The regressor is based on the design of CrystalNet, with a densely connected architecture with one-dimensional input and output. Specifically, for a given depth in the network,DenseNet aggregates previous intermediate data representations as input to the next convolutional layer.As shown in the figure below.
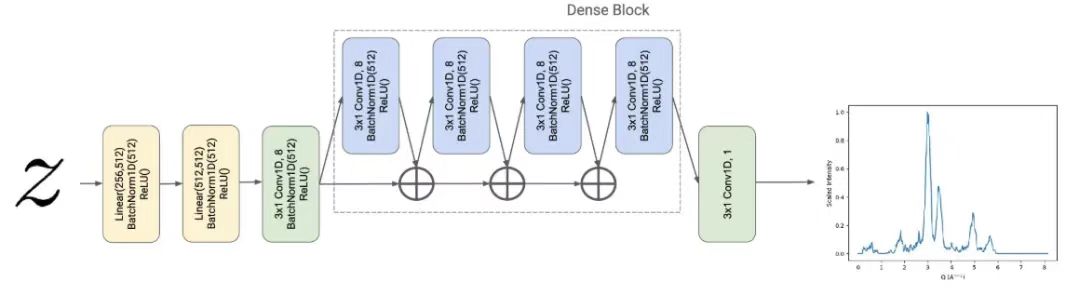
Research has shown that DenseNet reduces the vanishing gradient problem and achieves excellent results on standard computer vision benchmarks.
Experimental results: potential for real-world application
Typically, nanostructures are defined as crystals with a size less than 1000 Å, but to test the effectiveness of the proposed method, the researchers reduced the size of the crystals by two orders of magnitude, using a mathematical filtering method based on Fourier analysis to simulate the PXRD method for crystals with a size of 10 Å and 100 Å. As expected,The 10 Å case shows more peak broadening than the 100 Å case, indicating that the information degradation is more confirmed.As shown in the figure below.
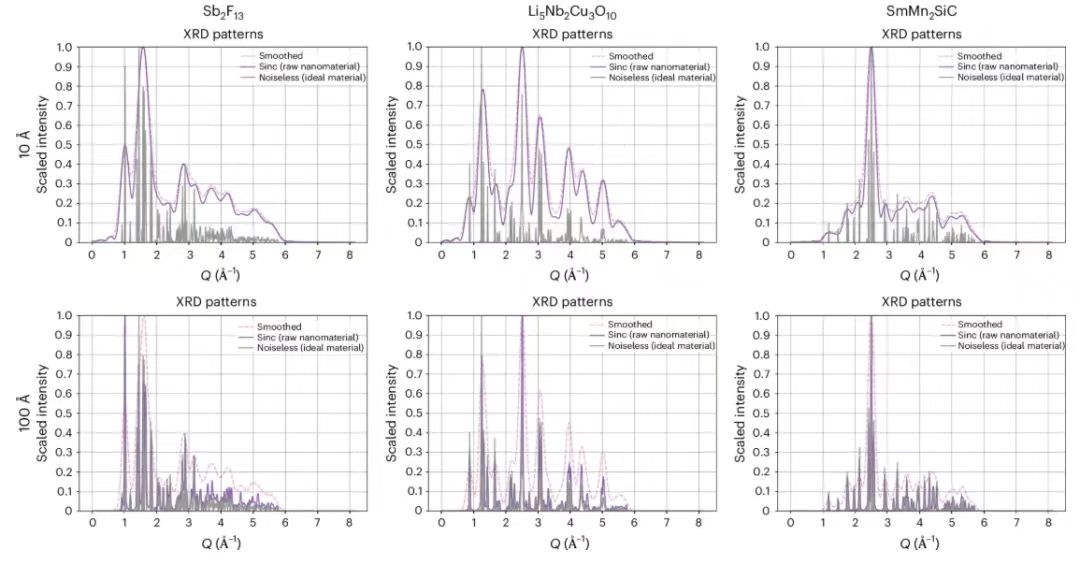
This figure shows how researchers simulated the effect of nanoscale shrinkage on PXRD peaks using sinc² filtering. The gray line represents the ideal mode, and the purple line represents the PXRD peak that is broadened after processing.To improve the model performance, the researchers further applied an additional Gaussian filter after the sinc filter.Although this will increase the broadening of the diffraction peak, it can effectively eliminate the sharp ripples caused by filtering. The horizontal axis represents the size of the scattering vector in Å⁻¹, and the vertical axis is the scaled diffraction intensity, where 1 represents the maximum intensity value.
Next, the researchers showed the PXRDnet structure predictions, as shown in the figure below. The leftmost column shows the real crystal structure, and the other columns show the reconstructed crystal structures of nanocrystals with diameters of 10 Å and 100 Å simulated by PXRDnet in the PXRD pattern, and after Rietveld refinement correction.
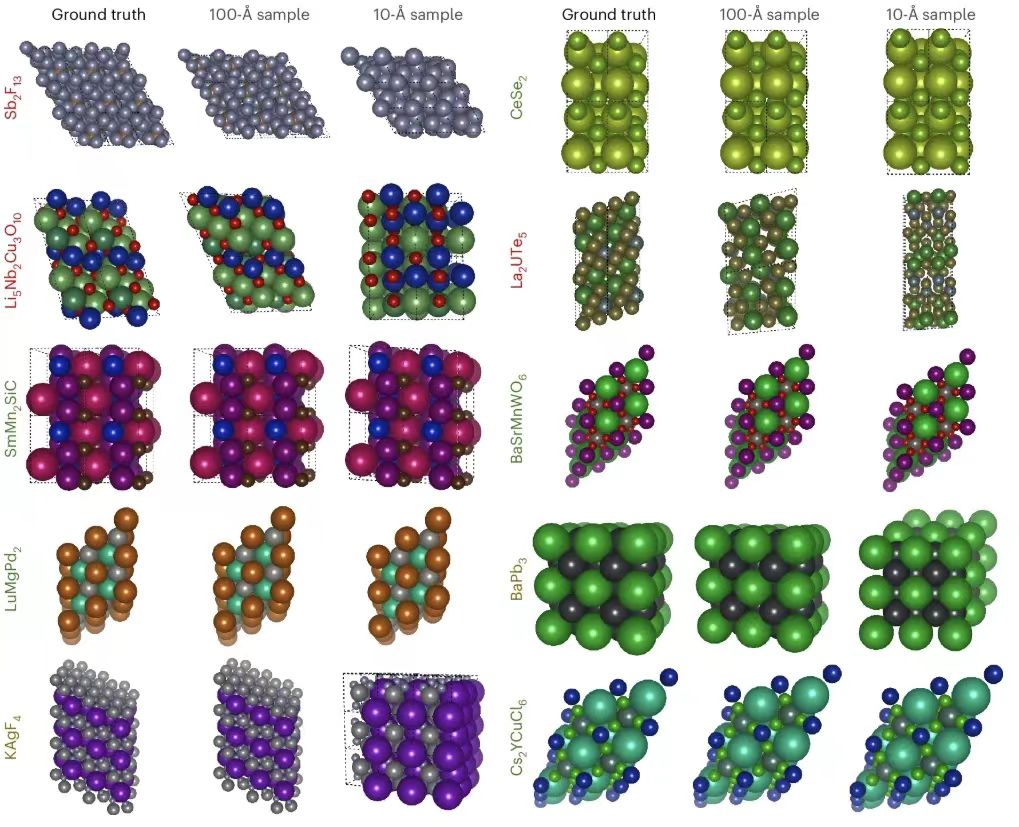
The results show that PXRDnet performs well in material structure analysis of various inorganic chemical compositions.The performance is slightly better at the 100 Å simulation crystal size, but remains excellent at the more challenging 10 Å simulation crystal size.For example, PXRDnet can successfully capture the crystal shape of materials such as Cs₂YCuCI₆ and SmMn₂SiC, and also successfully capture the symmetry of materials such as Cs₂YCuCI₆ and BaSrMnWO₆. In addition, even in some extreme cases, such as failure of Li₅Nb₂Cu₃O₁₀ or Sb₂F₁₃, PXRDnet can still provide valuable reference for experiments.
The figure below shows the comparison of the real PXRD pattern, the original predicted pattern by PXRDnet, and the pattern after Rietveld refinement, showing the degree of harmony between the predicted model and the real data, and verifying the necessity of Rietveld, which can effectively improve the prediction accuracy of the model. For example, at 100 Å, the predicted difference of Sb₂F₁₃ is 0.681, and after refinement (AI+Rietveld), it reaches 0.019.
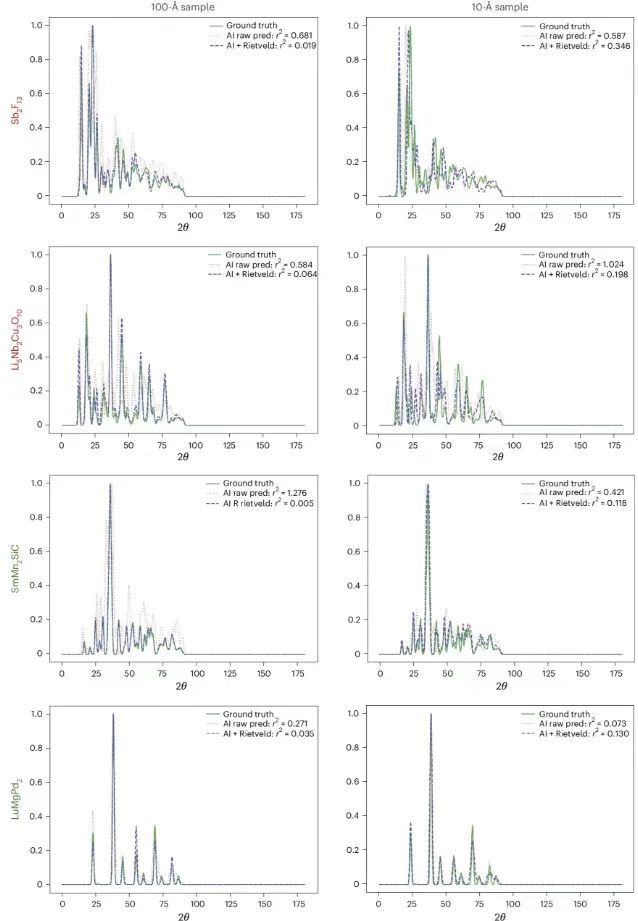
Comparison of the true PXRD pattern, the original PXRDnet predicted pattern, and the pattern after Rietveld refinement
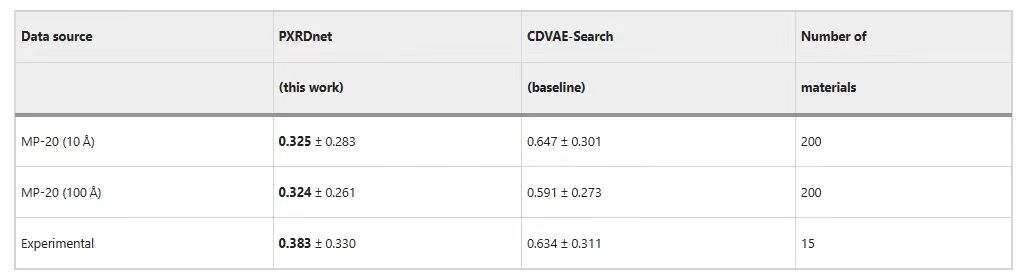
The following table shows that PXRDnet can successfully reconstruct the materials in MP-20.Compared with the CDVAE-Search baseline, the prediction results of PXRDnet are more outstanding.
To further improve the results, the researchers performed Rietveld refinement on 20 uniformly selected structures resolved by PXRDnet, selecting the top 10 candidate inputs for each structure, as shown in the figure below.
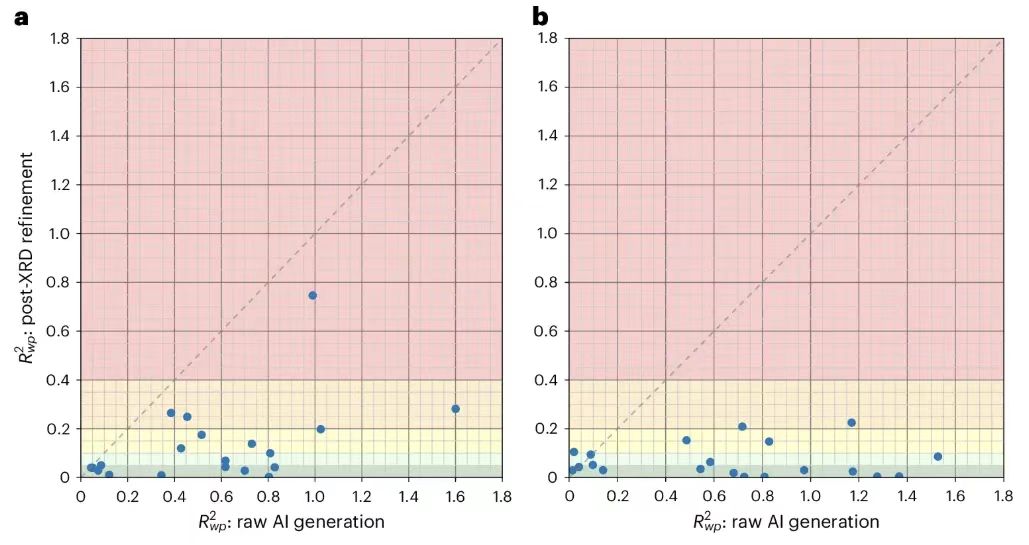
The results show thatRietveld refinement was particularly effective for the 100 Å tests, which have sharper Bragg peaks, with 18 of the 20 structures tested falling below 20% and 15 falling below 10%.This shows that despite some minor issues, PXRDnet is still able to consistently output results that are close to the true structure, and that the correct structure can be obtained with appropriate human intervention in each case.
Finally, the researchers verified the PXRD test performance through experiments, and the data came from the IUCr database, as shown in the figure below.
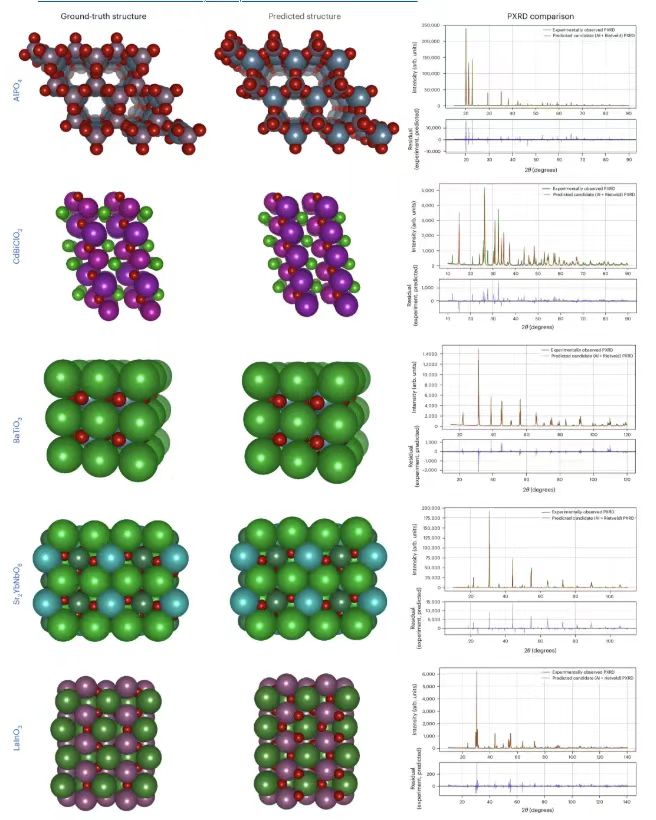
The leftmost column shows the benchmark structures, based on the experimentally observed PXRD patterns obtained from the IUCr database, the middle shows the structures predicted by PXRDnet, and the right side shows the comparison between the TOPAS (v.7) simulated PXRD and the actual experimentally observed PXRD.The results show that PXRDnet overcomes the simulation-to-reality gap and its results are comparable to those obtained from simulated data in terms of visual analysis and quantitative metrics, demonstrating the potential of the proposed model for application in real-world scenarios.
AI and materials science combine to solve century-old problems
The introduction of PXRDnet solves a century-long problem in the materials science community. As the paper states, this method, like any structural solution, is not 100% successful, but it provides a candidate method for exploring structural analysis, thus opening more doors to success.
Of course, the success of PXRDnet did not happen overnight, but was the result of continuous exploration by standing on the shoulders of giants. In the intersection of artificial intelligence and nanomaterials, countless researchers are constantly working on breakthroughs.
For example, the research published by MIT, Stanford University and other teams on "Crystal Structure Determination from Powder Diffraction Patterns with Generative Machine Learning"Here we present a groundbreaking generative machine learning model that can solve crystal structures from real experimental PXRD data.In the experiments, the researchers predicted the structures of 134 experimental patterns from the RRUFF database and thousands of simulated patterns from the Materials Project, with model matching rates reaching the state-of-the-art 42% and 67%, respectively.
Paper address:
https://pubs.acs.org/doi/10.1021/jacs.4c10244
In addition, teams from the Chinese Academy of Sciences, Shanghai Jiaotong University, Tsinghua University, and Renmin University of China have also published related research.We propose an end-to-end neural network, PXRDGen, that can determine crystal structure by learning the structural distribution of experimentally stable crystals and their PXRD patterns.The atomic precision structure is extracted from PXRD data. The model inherits a pre-trained XRD encoder, a diffusion/flow-based structure generator, and a Rietveld refinement module, and can accurately achieve structural analysis in just a few seconds. The related research was published under the title "Powder Diffraction Crystal Structure Determination Using Generative Models".
Paper address:
https://arxiv.org/abs/2409.04727
In summary, the exploration of PXRDnet and other methods has enabled the materials science community to move from traditional methods to the cross-integration of artificial intelligence and materials science. It has not only achieved substantial breakthroughs and solved the problems faced by the materials science community, but also provided new ideas and methods for subsequent research, injecting new vitality into the future development of materials science.








