Command Palette
Search for a command to run...
David Baker's team's Latest Research Uses Protein Sequence Generation Models to Achieve Overlapping Gene Design With a Very High Success Rate
In 1977, British biochemist Frederick Sanger first discovered a phenomenon that subverted cognition when analyzing the ΦX174 bacteriophage genome: the total length of proteins encoded by this 5.4kb DNA molecule far exceeded its physical capacity limit. The sequencing results revealed thatTwo pairs of genes share the same DNA region through different reading frames - this phenomenon is called overlapping genes (OLG) and is extremely common in the viral world.For example, in the 3.2kb genome of hepatitis B virus, the 50% region is covered by multiple pairs of overlapping genes, and more than half of the known viruses contain at least one OLG.
This breakthrough in intuitive genome design hides the survival wisdom of the virus: when the virus competes for limited space in the host cell, OLG uses the "gene stacking" strategy to allow a single nucleotide to participate in the encoding of two codons at the same time, achieving functional superposition in a compact sequence. The discovery of the Sanger team has opened up related research. Subsequent studies have shown that the proteins encoded by OLG often have high sequence degeneracy, and their amino acid sequence tolerance allows two functional proteins to coexist in the same DNA chain. More importantly, even proteins that need to form a clear three-dimensional structure can achieve folding compatibility in different reading frames through sequence arrangement.
However, the core question remains: Under the standard genetic code, can the degeneracy of amino acid sequences support the folding of any functional protein pair in overlapping frameworks? When nucleotides need to take into account dual coding, is the sequence space for protein folding severely restricted?
David Baker's team at the University of Washington recently used advanced generative models to conduct synthetic OLG design research and verify its feasibility from an engineering perspective.The research team designed overlapping sequences for two protein families to encode highly ordered de novo designed protein structures. Both computer simulation and experimental verification showed a very high success rate: under overlapping constraints, alternative reading frames can not only accommodate clear three-dimensional folding, but also their structural stability and functional integrity are comparable to those of non-overlapping sequences.
The relevant research results have been published as a preprint on bioRxiv under the title "Design of overlapping genes using deep generative models of protein sequences".

Paper address:
https://doi.org/10.1101/2025.05.06.652464
The open source project "awesome-ai4s" brings together more than 100 AI4S paper interpretations and provides massive data sets and tools:
https://github.com/hyperai/awesome-ai4s
Dataset: Integrating multi-dimensional data resources and analysis methods
In order to analyze the plasticity of genetic code and its application in protein design, this study integrates multi-dimensional data resources and analysis methods to construct a complete research chain from theoretical design to experimental verification.
In genetic code randomization studies,The study generated 1,000 alternative codon combinations based on amino acid permutation and codon shufflers strategies.This dataset ensures sample diversity and uniformity through clear algorithm design, providing a statistical benchmark for evaluating the functional impact of codon rearrangements.
At the same time, the study selected 3 representative secondary structure target proteins and constructed 9 paired combinations, which achieved standardization of experimental conditions under the premise of controlling variables and effectively connected the correlation analysis between genetic code variation and protein structure function.
In the protein domain sequence analysis phase, the study extracted seed sequences from the Pfam 37.0 database, randomly sampled sub-regions of 100 amino acids in length, and used the Markov model to generate synthetic protein sequences that retained the k-mer distribution.This method combines bioinformatics screening and statistical modeling, which not only retains the sequence characteristics of natural proteins, but also creates control samples by introducing controllable random variables.It provides an innovative dataset that combines natural properties with artificially designed features for subsequent analysis.
In the protein language model embedding analysis, the researchers extracted the hidden layer features of ESM2, ESM3 and ProstT5, and projected them into two-dimensional space through the UMAP algorithm after position averaging. By accurately setting parameters such as n_neighbors = 15, the high-dimensional sequence features were converted into intuitive topological maps.While preserving the sequence similarity structure, it provides a unified visualization framework for cross-model comparison.It demonstrates the cutting-edge combination of computational biology and data visualization.
During the experimental verification phase,The researchers cloned and recombined 192 overlapping genes to generate 384 frame-shifted protein variants.The experiment strictly controlled key parameters: 20 hours of culture at 37°C ensured the stability of the E. coli expression system, and the 6M guanidine hydrochloride gradient renaturation scheme ensured the correct folding of inclusion body proteins. This quantitative control of the entire process from molecular design to purification characterization not only improved the repeatability of research conclusions, but also provided a standardized experimental paradigm for protein engineering.
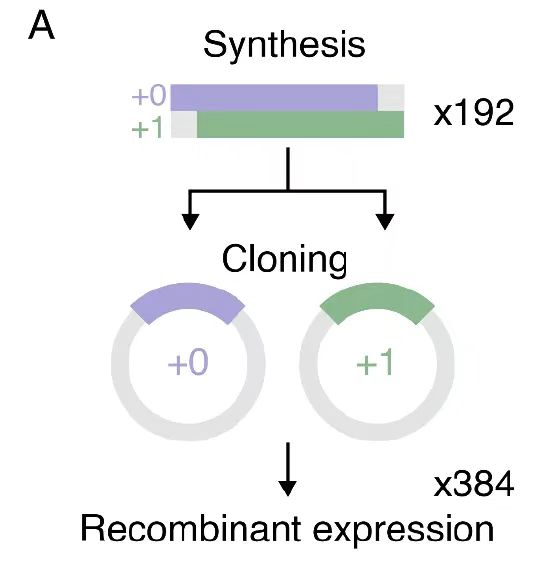
OLG design based on generative model: a multi-framework compatible sequence synchronization optimization method
This study developed a computational algorithm that effectively addresses the problem of sequence space constraints caused by the interdependence of coding frames in overlapping gene (OLG) design, and achieved simultaneous optimization of the adaptability of two protein sequences.
At the algorithm design level, the study integrated generative models such as EvoDiff-MSA and ProteinMPNN.The former is based on the MSA Transformer architecture and can generate design sequences based on the target protein multiple sequence alignment (MSA) through autoregressive diffusion target training; the latter is a structure-conditional generation model that can design corresponding protein sequences given a three-dimensional structure. Both models use position-by-position masking and constrained sampling strategies to generate overlapping sequence libraries covering a variety of offsets and framework arrangements.
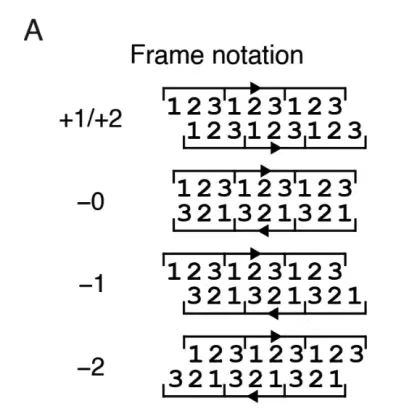
As shown in Figure A below, this study proposed a frame-by-frame iterative sampling strategy for the phase constraints of five variable reading frames (+1, +2, -0, -1, and -2).
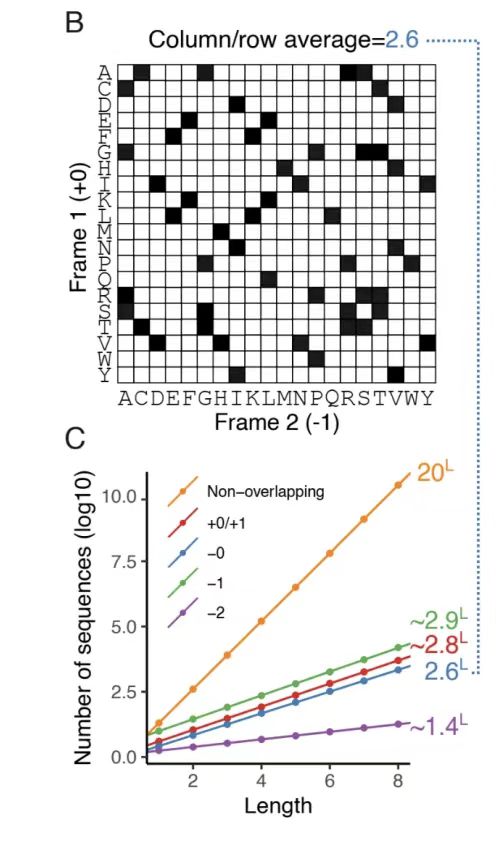
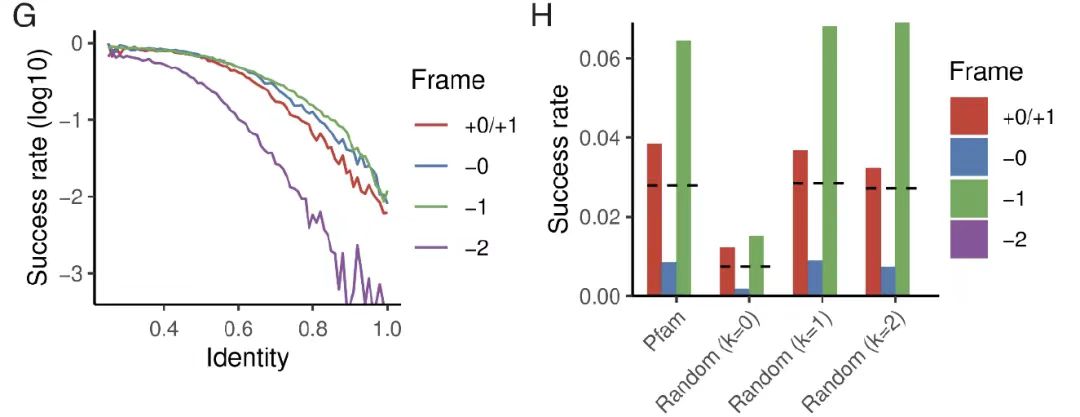
As shown in Figure B below, by analyzing the amino acid compatibility matrix of the -0 framework, it was found that there are an average of 2.6 compatible amino acid choices for a single position in the reference framework, forming 52ⁿ (n is the sequence length) potential overlapping sequence pairs, highlighting the design space brought by the degeneracy of the genetic code. The degrees of freedom of other frameworks were quantified with the help of Monte Carlo approximation, as shown in Figure C below. The results show that the +1 and -1 frameworks have higher degrees of freedom (about 2.8 and 2.9, respectively), while the -2 framework has a significantly limited degree of freedom (about 1.4) due to the low efficiency of codon degeneracy.
Finally, as shown in Figure D below, the algorithm systematically scans the sequence positions (Scan order) and dynamically updates the joint probability matrix in each scan in combination with the adjacent amino acid constraints.After multiple rounds of iterations, the generated overlapping sequence pairs are ensured to meet the compatibility of the framework.This strategy can be extended to complex frameworks with phase offsets, optimizing the design quality by biasing the scan order and providing key constraints for iterative decoding of the generative model.
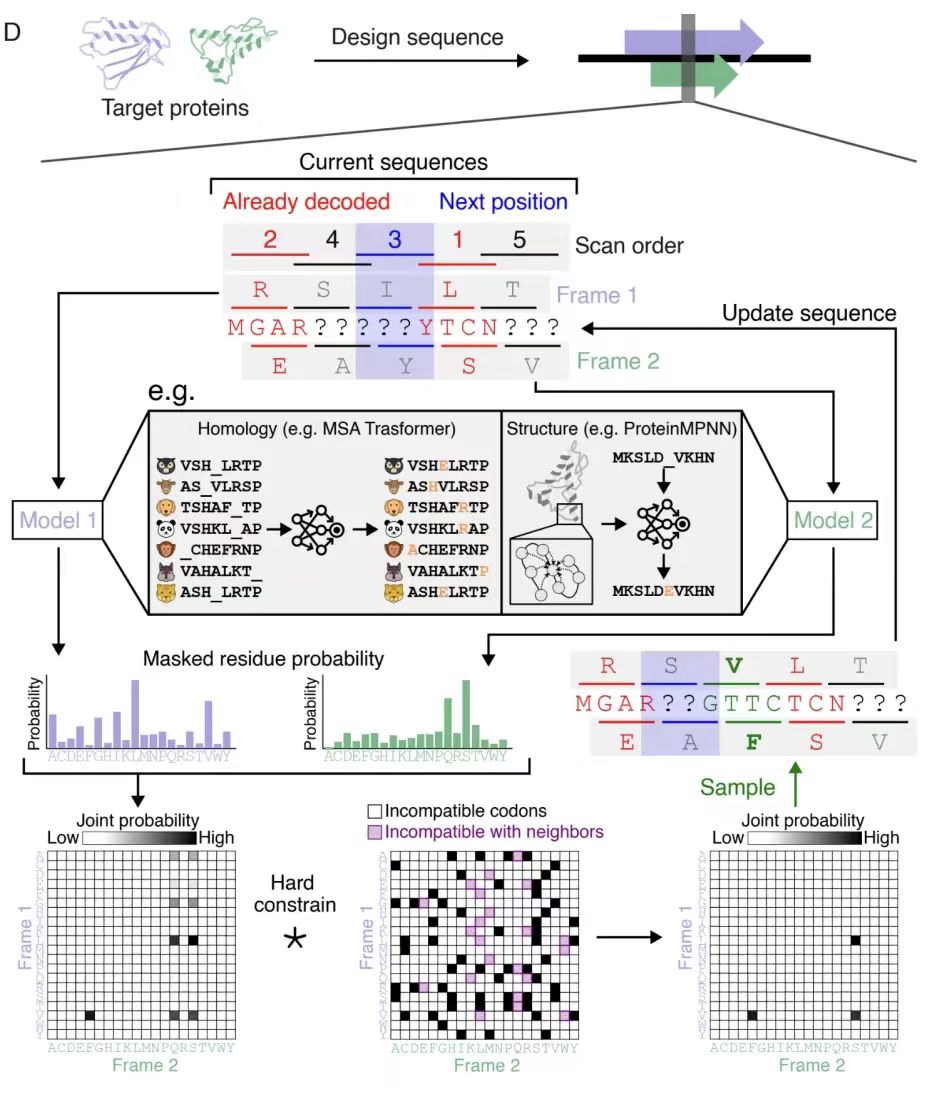
Beyond the natural template limitation: efficient generation of synthetic OLGs of arbitrary protein pairs
The experimental design covers multiple directions, including homology-based OLG design evaluation, overlapping feasibility analysis of highly ordered protein main-chain structures, evolutionary accessibility studies of OLG sequences, and experimental verification.
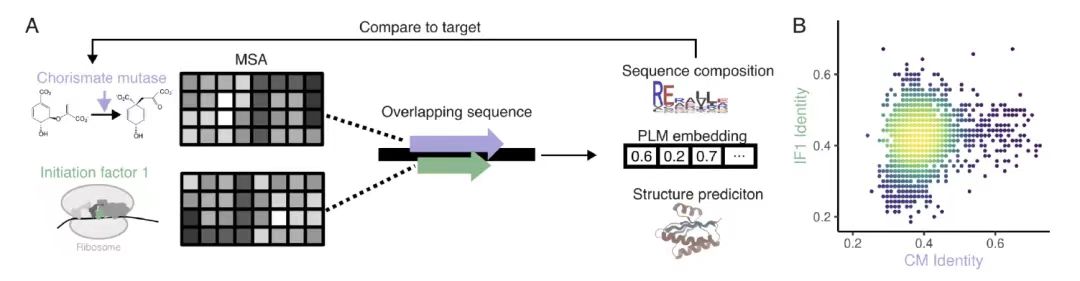
In the homology-based OLG design evaluation,As shown in Figure A below, the research team selected bacterial shikimate mutase (CM) and translation initiation factor 1 (IF1) as targets, used the EvoDiff-MSA generation model, and used multiple sequence alignment (MSA) as conditional context to generate 3,307 completely overlapping sequence designs through position-by-position masking and constrained sampling.
As shown in Figure B below, although the homology between the designed sequence and the natural sequence is only 38.9% (CM) and 42.3% (IF1),However, the protein language model embedding analysis shows that its distribution in two-dimensional space is highly consistent with the natural sequence.This indicates that these designed sequences are credible members of the target protein family, validating the algorithm's design capabilities for natural protein families.
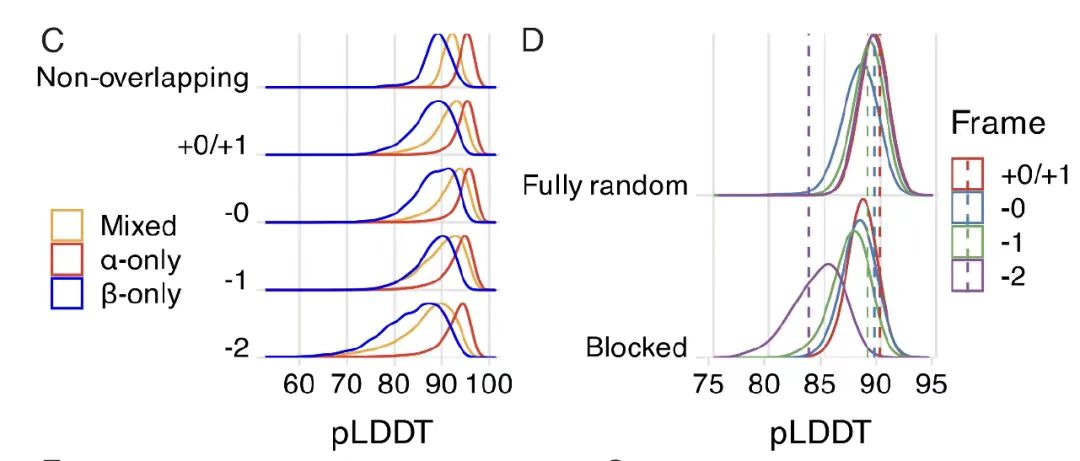
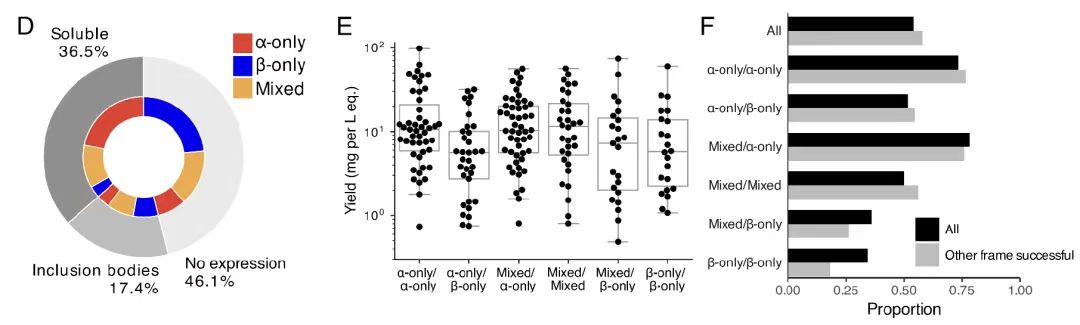
When exploring the feasibility of overlapping highly ordered protein backbone structures,As shown in Figure A below, researchers used the ProteinMPNN structure conditional generation model to generate 56,250 overlapping designs and 33,000 non-overlapping designs for 15 de novo generated main chain structures (covering α, β and mixed folding categories). As shown in Figure B below, the AlphaFold2 evaluation data shows thatThe average pLDDT value for the overlapping design was 90.2, which was close to the 92.0 for the nonoverlapping design.
Further analysis revealed that, as shown in Figure CD below, only the -2 frame performed poorly due to the low efficiency of codon degeneracy. Randomized genetic code analysis showed that the natural genetic code (SGC) had a significant advantage in encoding OLG, and performed well except for the -2 frame, and had a composition preference for highly degenerate amino acids.The mechanism by which SGC structure affects the feasibility of overlapping sequences was revealed.
In evolutionary accessibility studies,The research team started with a seed protein sequence with a fixed number of mutations. As shown in the figure GF below,The study found that even under the extreme condition of zero mutation, about 1% designs can still achieve high structural stability (pLDDT>85, TM>0.7);When natural Pfam sequences were used as parents, the success rate increased to 3%, and this result was consistent with random sequences that retained first-order compositional deviations. This fully demonstrates that highly optimized natural proteins can accommodate new proteins in alternative frameworks without major sequence changes, verifying the feasibility of OLG at the evolutionary level.
In the final experimental verification, the research team recombined and expressed 192 overlapping sequences and characterized their structures. The results showed that, as shown in Figure B,Individual proteins of 54% were successfully expressed, and most had the expected secondary structures and high thermal stability.
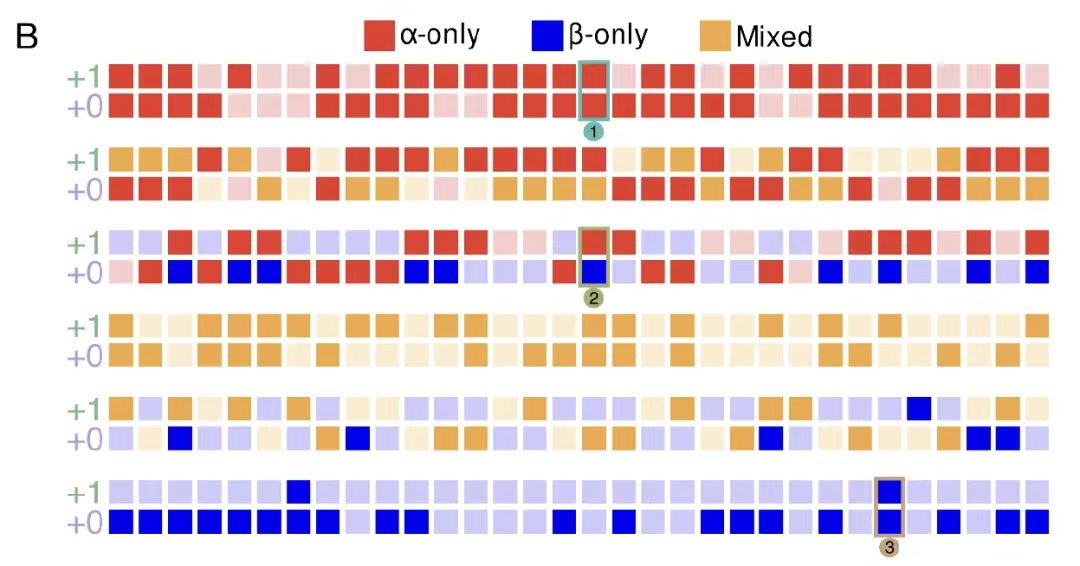
Furthermore, as shown in Figures DF below, the success rate varied depending on the secondary structure content of the protein, with α-helical proteins having the highest success rate. In addition, overlapping pairs of 31% were successfully purified, and the success of one framework did not affect the success of the other framework.These results further support the high feasibility and experimental validation rate of the OLG sequences, demonstrating the effectiveness of the algorithm in designing functional and structurally stable overlapping proteins.
Frontier exploration in the field of synthetic biology, OLG engineering applications gradually deepen
In the field of synthetic biology, research teams and companies in many parts of the world are engaged in in-depth exploration of the engineering applications of overlapping genes (OLGs).
For example, Zhu Ting's research group from Tsinghua University has made significant progress in the study of mirror biological systems and successfully achieved the fully chemical synthesis of mirror Pfu DNA polymerase.This not only makes the assembly of mirror DNA of kilobase length a reality, but also develops information storage technology based on mirror DNA.This technology uses the coding strategy of mirror genes to provide a new idea for the bidirectional functional superposition of OLG. When the double helix structure of mirror DNA carries both natural and mirror genetic information, the utilization rate of sequence space is significantly improved, providing an important foundation for the compact design of artificial genomes.
* Paper link:https://www.nature.com/articles/s41587-021-00969-6
In addition, Christopher Voigt's team at the Massachusetts Institute of Technology has developed a synthetic biology platform based on gene circuit design. They have successfully achieved modular assembly of metabolic pathways by reconstructing the regulatory logic of prokaryotic gene clusters. This technical approach is closely aligned with OLG's design philosophy.When multiple functional genes form a compact genetic module through overlapping sequences, it can not only reduce the redundancy of the genome but also improve the stability of the system through coordinated expression.For example, the artificial nitrogen-fixing gene cluster designed by the team adopted the OLG strategy to compress the coding sequences of multiple key enzymes into the same DNA region, significantly reducing the metabolic burden of the host cells while ensuring catalytic efficiency.
* Paper link:https://www.nature.com/articles/s41467-022-33272-2
It is worth noting that these studies not only reveal the widespread existence of OLG in natural evolution, but also verify its biophysical feasibility through engineering means. In the study introduced in this article, David Baker's team used a deep learning model to design a synthetic OLG, which showed structural stability comparable to that of the natural sequence in computer simulations. The high success rate of experimental verification further proves the biological compatibility of overlapping coding. This complete closed loop from basic research to application transformation is reshaping the design logic of synthetic biology, and is expected to bring new breakthroughs in many fields such as innovative drug development, precision diagnosis, and cell therapy.
References:
1.https://www.tsinghua.edu.cn/info/1181/86148.htm
2.https://tech.huanqiu.com/article/9CaKrnJUV0x
3.https://news.bioon.com/article/4161e88572ad.html








