Command Palette
Search for a command to run...
The Westlake University Team open-sourced SaProt and Other Protein Language Models, Covering Structure Function prediction/cross-modal Information search/amino Acid Sequence Design, etc.

On March 22-23, 2025, the "AI Protein Design Summit" of Shanghai Jiao Tong University was officially held.The summit brought together more than 300 experts and scholars from well-known universities such as Tsinghua University, Peking University, Fudan University, Zhejiang University, and Xiamen University, as well as more than 200 representatives from leading industry companies and technical R&D personnel, to discuss in depth the latest research results, technological breakthroughs, and industrial application prospects of AI in the field of protein design.

During the summit,Dr. Yuan Fajie from Westlake University shared the latest research progress of protein language models with the theme of "Research and Application of Protein Language Models" and introduced the important achievements of the team in detail.Including protein language models SaProt, ProTrek, Pinal, Evolla, etc. HyperAI has compiled and summarized their in-depth sharing without violating the original intention. The following is a transcript of the highlights of the speech.
A protein language model worth noting
Proteins are biological macromolecules composed of 20 amino acids connected in series. They perform key functions in the body, such as catalysis and metabolism, and are the main executors of life activities. Biologists usually divide the structure of proteins into four levels: the primary structure describes the amino acid sequence of the protein, the secondary structure focuses on the local conformation of the protein, the tertiary structure represents the overall three-dimensional configuration of the protein, and the quaternary structure involves the interaction between multiple protein molecules.In the field of AI protein, research is mainly based on these structures.
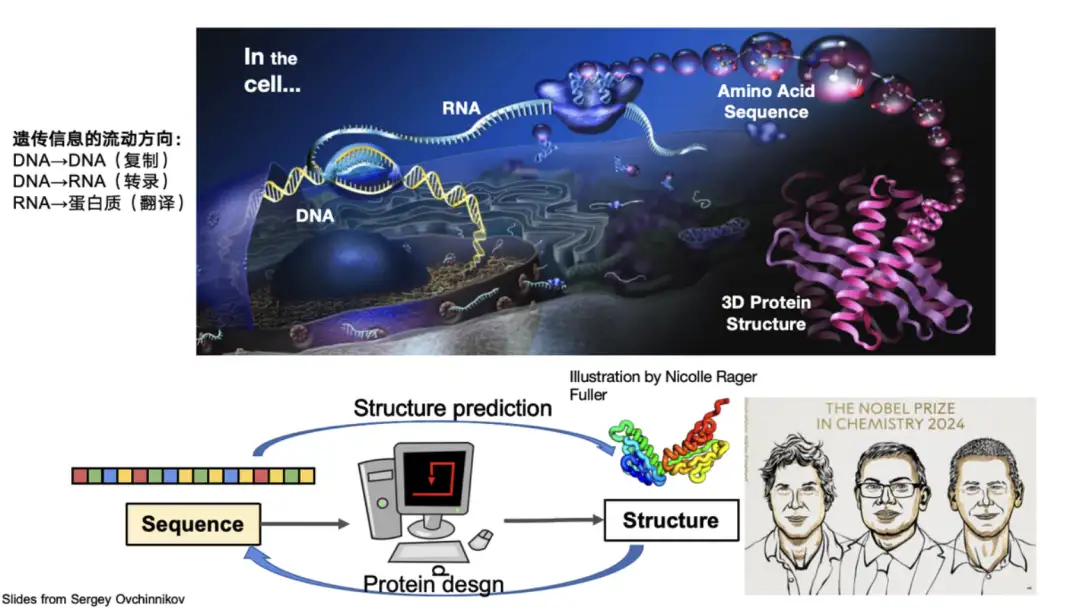
For example, predicting the three-dimensional structure of a protein from its sequence is the core problem solved by AlphaFold 2. It solved the protein folding problem that has troubled the scientific community for 50 years and won the Nobel Prize for it. Conversely, Professor David Baker, an important contributor to the field of protein design, which designs new protein sequences based on structure and function, also won the Nobel Prize.
Traditionally, protein structures are usually represented in the form of PDB coordinates. In recent years, researchers have explored methods to convert continuous spatial structure information into discrete tokens, such as Foldseek, ProTokens, FoldToken, ProtSSN, ESM-3, etc.
*Foldseek can encode the three-dimensional structure of a protein into one-dimensional discrete tokens.
Our team's protein language model is based on these discrete results.
Most AI + protein research can be traced back to natural language processing research, so let’s first review two classic language models in the field of natural language processing (NLP):One is the unidirectional language model represented by the GPT series,Its mechanism is based on the flow of information from left to right, predicting the next token based on the data on the left (above).One is the bidirectional language model represented by BERT,It is pre-trained through the Masked Language Model, which can see the information (context) on the left and right sides of a cooked word and predict the cooked word.
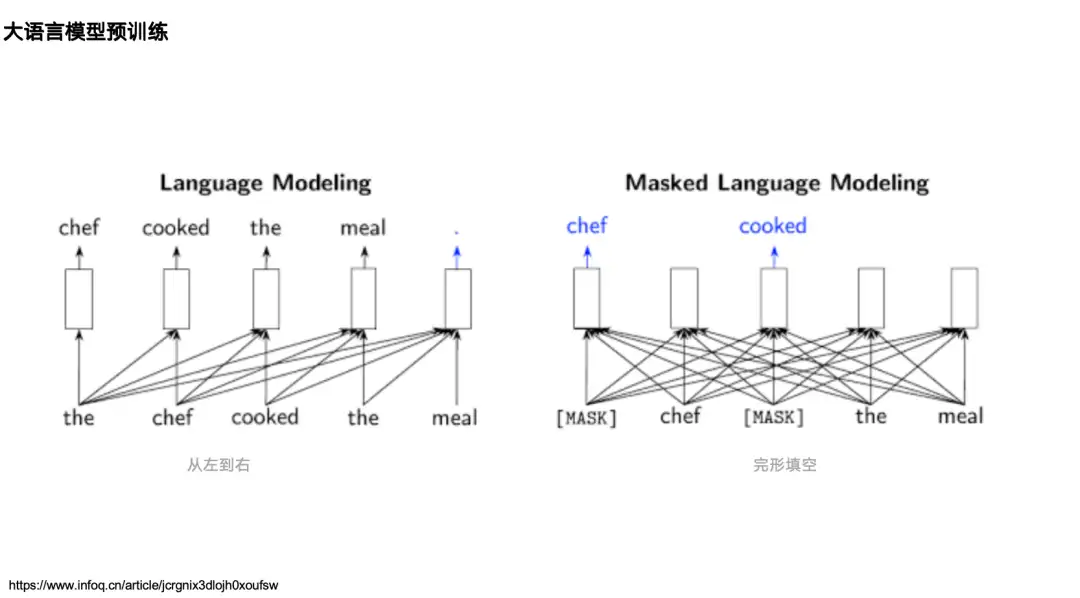
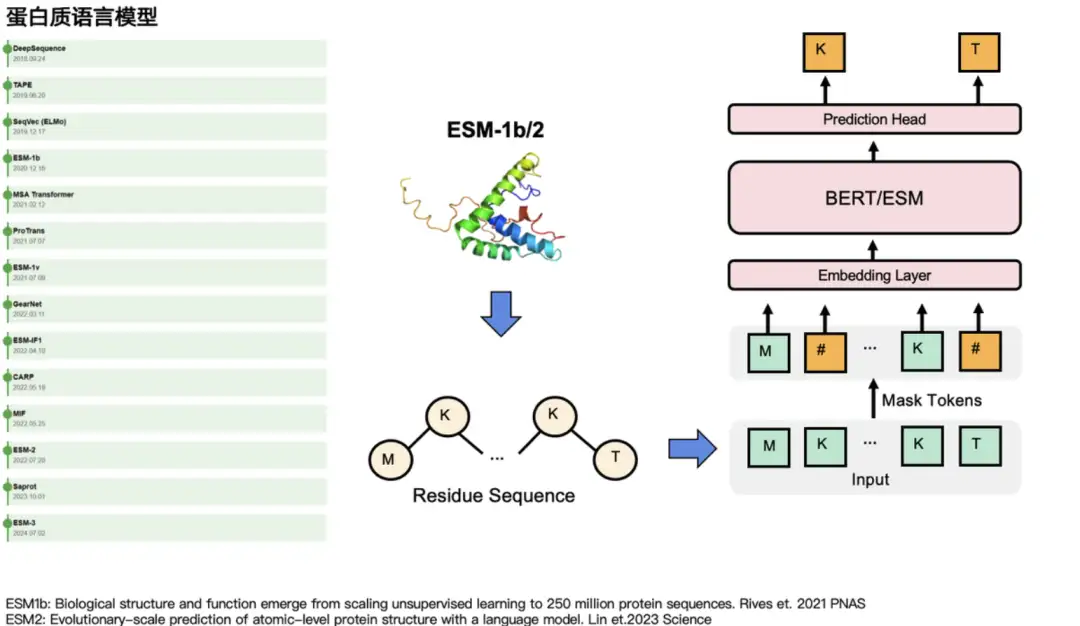
In the protein field, both types of models have their corresponding protein language models.For example, for GPT, there are ProtGPT2, ProGen, etc. For BERT, there are ESM series models: ESM-1b, ESM-2, ESM-3, which mainly mask some amino acids and predict their "true identity". In natural language tasks, they mask some words and then predict them. As shown on the left side of the figure below, other language models with great influence in the protein community include MSA Transformer, GearNet, ProTrans, etc.
Selected for ICLR 2024, protein language model SaProt integrates structural knowledge
The first result I want to introduce to you is SaProt, a protein language model with structure-aware vocabulary.This paper, titled "SaProt: Protein Language Modeling with Structure-aware Vocabulary", was selected for ICLR 2024.
In this paper, we proposed the concept of structure-aware vocabulary, combined amino acid residue tokens with structure tokens, and trained a large-scale universal protein language model SaProt on a dataset of approximately 40 million protein sequences and structures. This model comprehensively outperformed existing mature baseline models in 10 important downstream tasks.
SaProt open source address:
https://github.com/westlake-repl/SaProt
SaProt paper address:
https://openreview.net/forum?id=6MRm3G4NiU
Why do we make this model?
In fact, the input information of most protein language models is mainly based on amino acid sequences. After the breakthrough of AlphaFold, the DeepMind team cooperated with the European Bioinformatics Institute (EMBL-EBI) to release the AlphaFold Protein Structure Database, which stores 200 million protein structures. So we began to think: Can we integrate the structural information of proteins into the language model to improve its performance?

Our approach is very simple: we use Foldseek to convert the structural information of proteins from coordinate form to discrete tokens, thereby constructing an amino acid vocabulary and a structural vocabulary, and then cross-combining these two vocabulary to generate a new vocabulary, namely the structure-aware vocabulary (SA token). In this way, the original amino acid sequence can be converted into a new amino acid sequence - in this sequence, uppercase letters represent amino acid tokens and lowercase letters represent structural tokens, and then we can continue to work on the Masked Language Model. Based on this, we trained a 650 million parameter SaProt model, which used 64 A100 GPUs and took about 3 months to train.
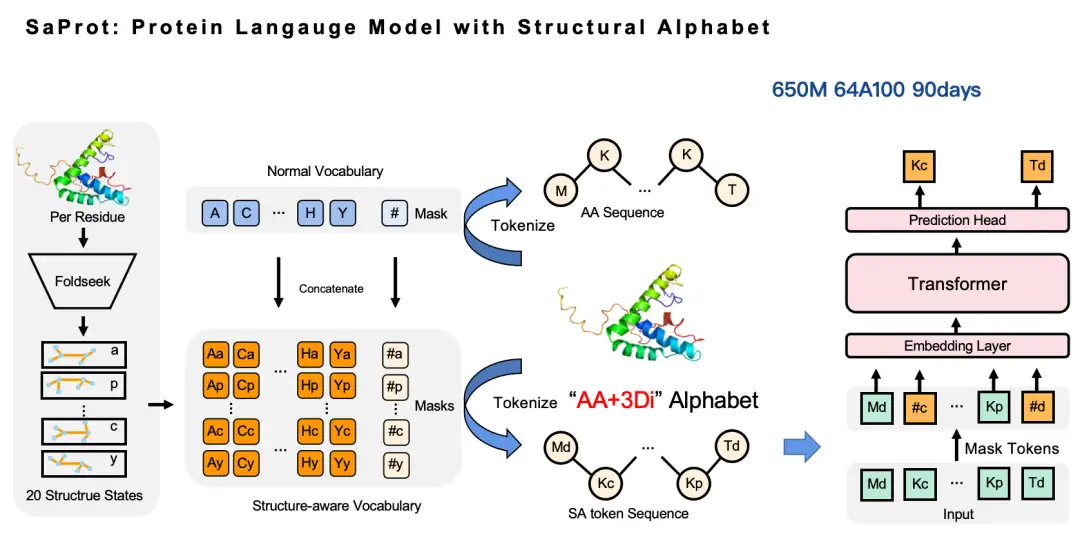
Why did we choose Foldseek to convert protein structure tokens?
We spent half a year exploring before we finally decided to use the Foldseek 3Di token sequence. Intuitively, incorporating structural information into the protein language model should improve performance, but in actual attempts, we tried many methods without success. For example, we used the GNN method to model protein structure. Because the protein structure is actually a graph neural network, we naturally want to model the protein structure as a graph, so we used the MIF method, but found that the trained model has poor generalization ability and cannot be extended to the real PDB structure. After in-depth analysis, we believe that this may be because the modeling method using the Masked Language Model will result in information leakage problems.
Simply put, the protein structure predicted by AlphaFold itself has some bias, pattern, and traces of AI prediction. When using these data to train a language model, the model can easily capture these traces, resulting in the model performing well on the training data but having very poor generalization ability.
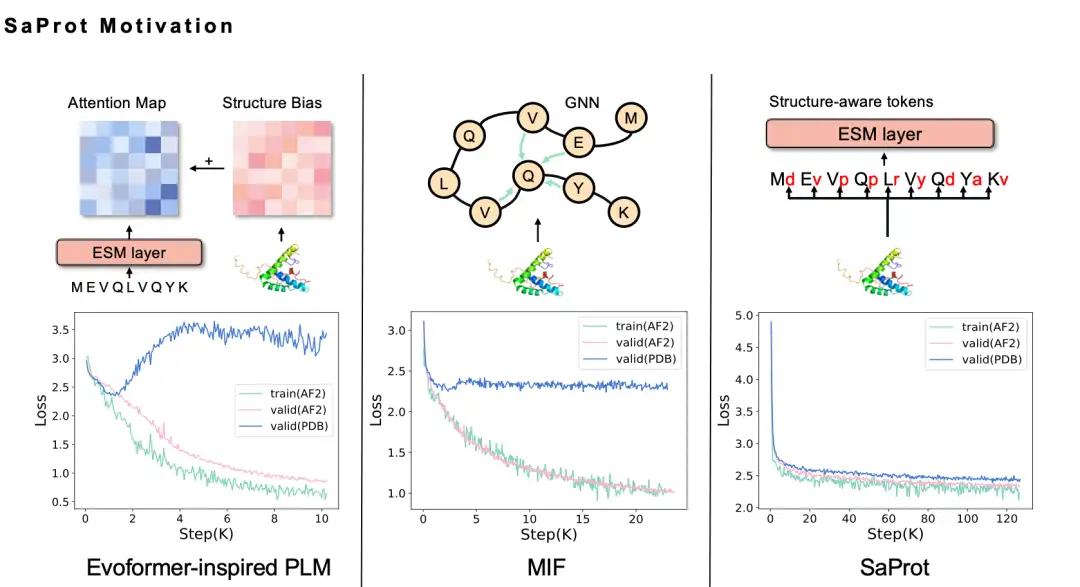
We tried various improvements, including using the Evoformer method, but the information leakage problem still existed until we tried Foldseek. We found that the loss of the SaProt model obtained on the structural data predicted by AlphaFold was reduced, and the loss on the real PDB structure data was also significantly reduced, which met our expectations.
In addition, SaProt performs well on multiple benchmarks.Last year, it was ranked first on the authoritative list ProteinGym. At the same time, we also collected the community's wet experiment verification of SaProt/ColabSaProt on more than 10 proteins (such as various enzyme mutations, fluorescent protein modification and fluorescence prediction, etc.), all of which performed well.
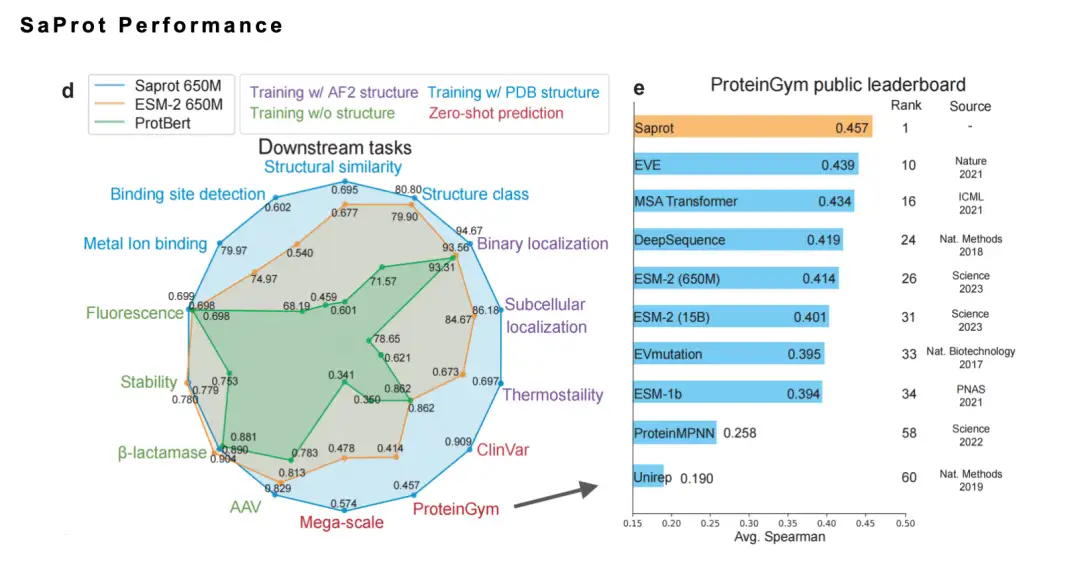
Although we think the SaProt model is pretty good,But considering that many biologists have not received training in deep learning,It is very difficult for them to independently fine-tune a protein language model with about 1 billion parameters.So we built an interactive interface platform ColabSaprot + SaprotHub.
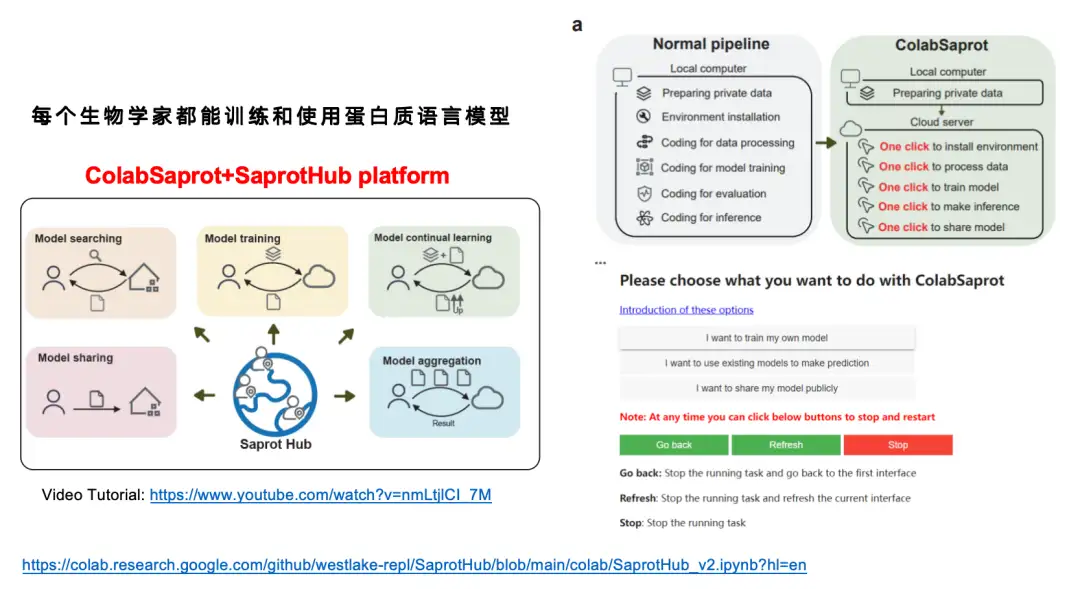
In the traditional model training process (Normal pipeline), users need to go through multiple steps such as data preparation, environment configuration, code writing, data processing, model training, model evaluation, model reasoning, etc. With ColabSaprot, the whole process is greatly simplified - users only need to click a few buttons to complete environment installation, model training and prediction, which greatly reduces the threshold for use.
As shown in the figure below, ColabSaprot mainly consists of three parts: training module, prediction module and sharing module.

* In the training module, users only need to describe the task on the left and upload the data, then click on training. The system will automatically select the optimal hyperparameters (such as Batch Size, etc.).
* In the prediction module, users can directly load their previously trained models and make predictions. They can also directly import models shared by other researchers to make predictions.
* The sharing module provides a way to protect data privacy while sharing results. The data of many laboratories are extremely valuable, and some researchers may need to use these data for follow-up research, but they still want to share existing models. In ColabSaprot, users can only share the model itself. Since the model is essentially a black box, others cannot obtain the original data.
When sharing models, considering that language models are usually large in size, it is almost infeasible to directly share a model with 1 billion parameters online.Therefore, we adopted a mature Adapter mechanism.Users only need to share a very small number of parameters, usually only 1% or 1/1,000 of the original model parameters. Everyone can share adapters with each other and load other people's adapters to make fine adjustments or predictions. If the improvement is good, new adapters can be shared again, thus forming an efficient community cooperation mechanism and greatly improving research efficiency.
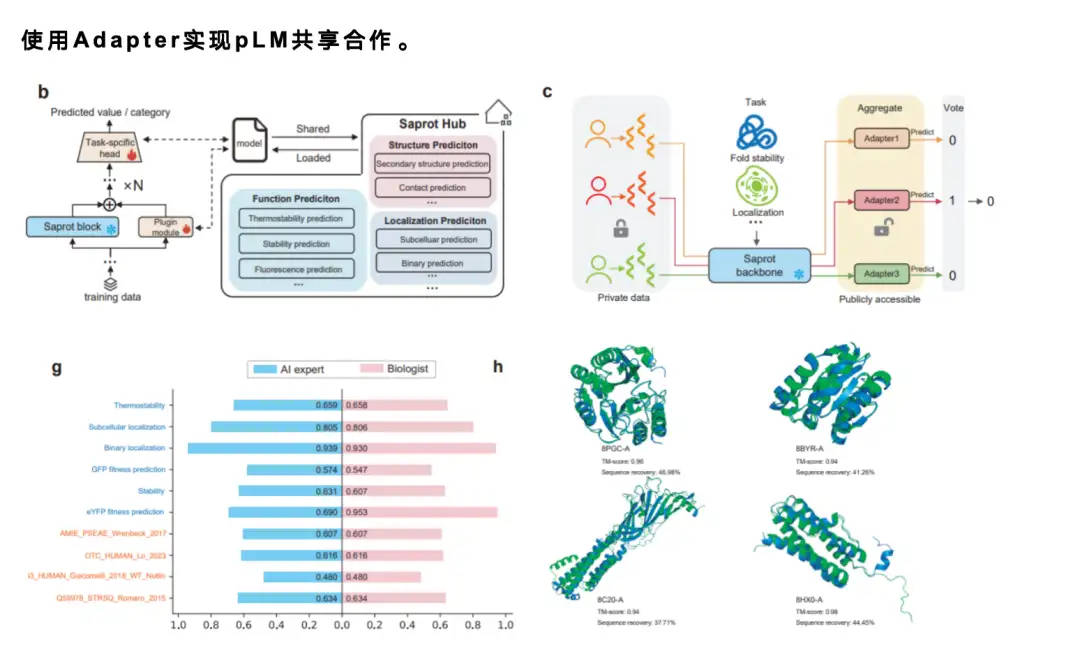
In addition, we also conducted a user study.We invited 12 students with no machine learning or programming background to try out the ColabSaprot platform. We provided them with data and told them the tasks to be done, and asked them to use ColabSaprot for model training and prediction. Finally, we compared their results with the performance of AI experts, and found that these non-professional users were able to reach a level close to that of experts after using ColabSaprot.
In addition, in order to promote the sharing of protein language models,We also set up a community called OPMC,Well-known scholars from home and abroad in this field participated in it, encouraging everyone to share models and promote cooperation and communication.
OPMC Address:

ProTrek model: Find the correspondence between protein sequence, structure and function
The second work I want to introduce is the protein language model ProTrek.
In biological research, many scientists are faced with this need: they have a genome with many proteins but do not know their specific functions.
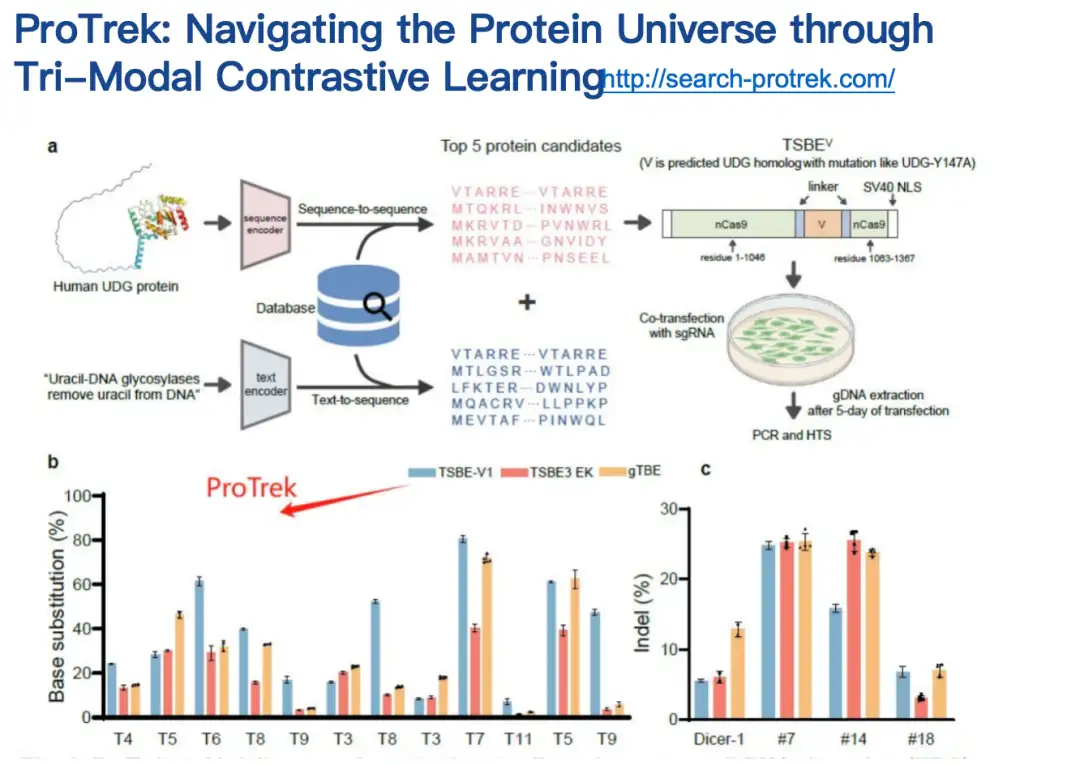
ProTrek is a trimodal language model for contrastive learning of sequence, structure, and function.With its natural language search interface, users can explore the vast protein space in seconds and search for relationships between all nine different tasks of sequence, structure, and function. In other words, with ProTrek, users only need to enter a protein sequence and click a button to quickly find information related to protein function and structure. Similarly, you can also find sequence and structure information based on function, and sequence and function information based on structure. In addition, it also supports sequence-sequence and structure-structure type searches.
ProTrek use address:
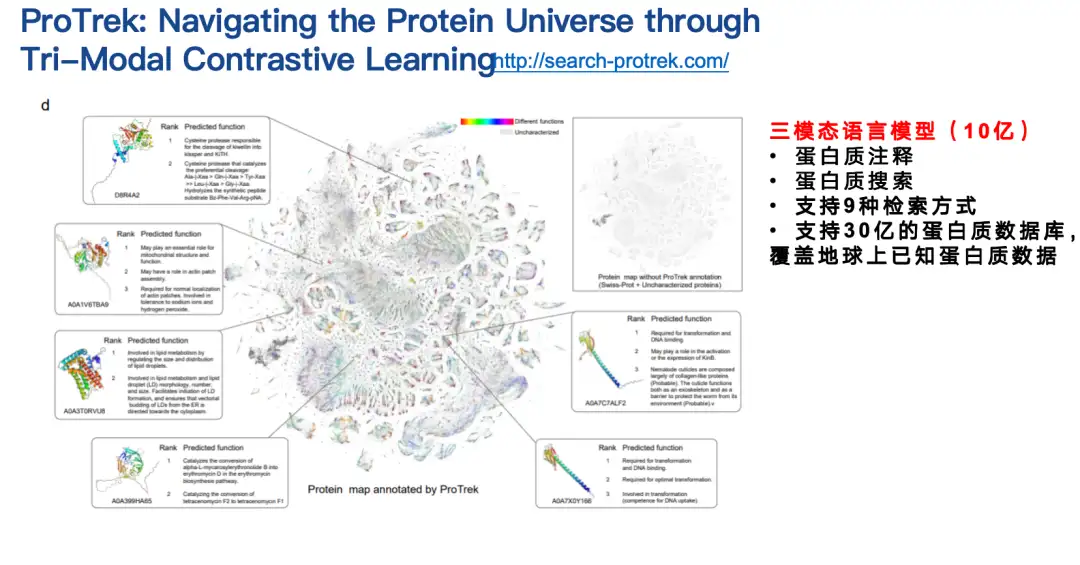
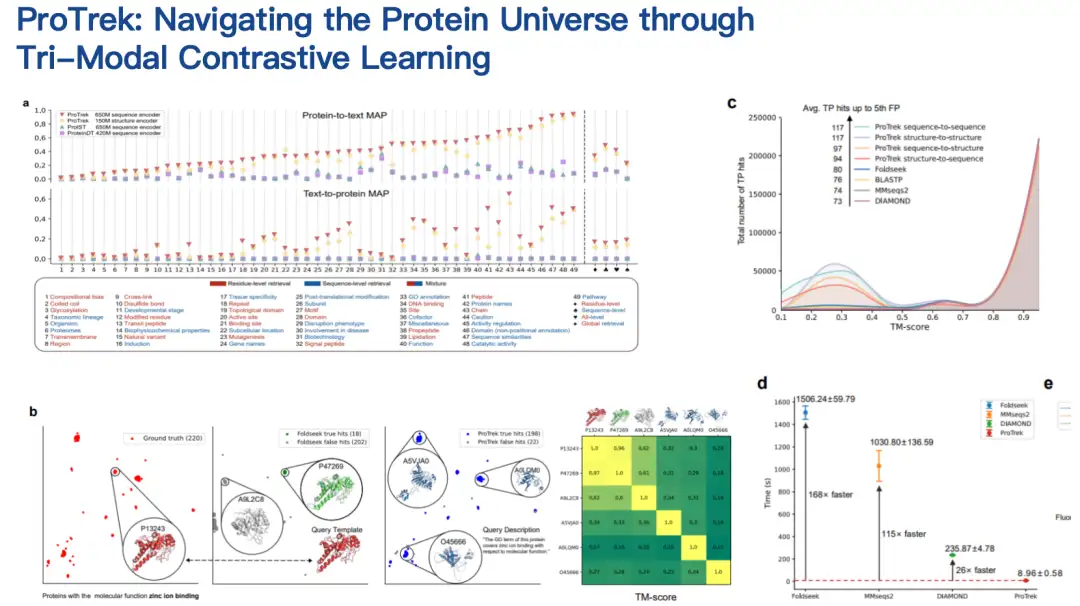
Our collaborators have evaluated the ProTrek model in both dry and wet tests.ProTrek achieves significant performance improvements over existing methods. We also use ProTrek to generate a large amount of data for training our generative models, which also performs well.
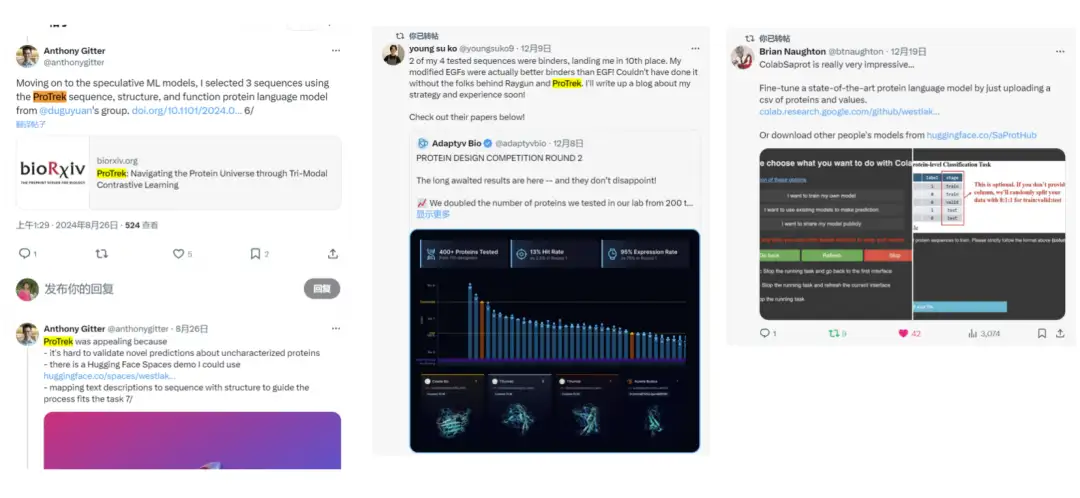
We noticed on Twitter thatMany users have started using ProTrek to compete.We also received a lot of positive feedback, which further proved the practicality of the model.
Pinal model: Design new protein sequences by simply inputting text
Another of our works is Pinal, a model for designing proteins based on text descriptions.
Traditional protein design usually needs to consider complex factors, such as biophysical energy function template information, etc. What we want to explore is, since large language models perform well in many tasks, can we design a text-based protein language model? In this model, we only need to simply describe the information of a protein to design its amino acid sequence?
Pinal use address:
http://www.denovo-pinal.com/
Paper address:
https://www.biorxiv.org/content/10.1101/2024.08.01.606258v1
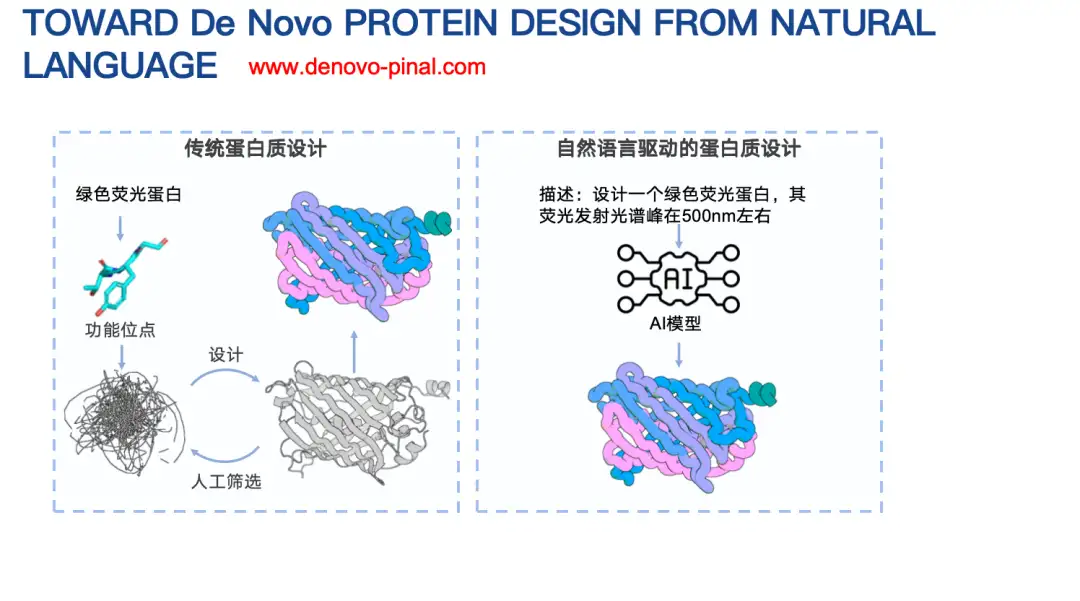
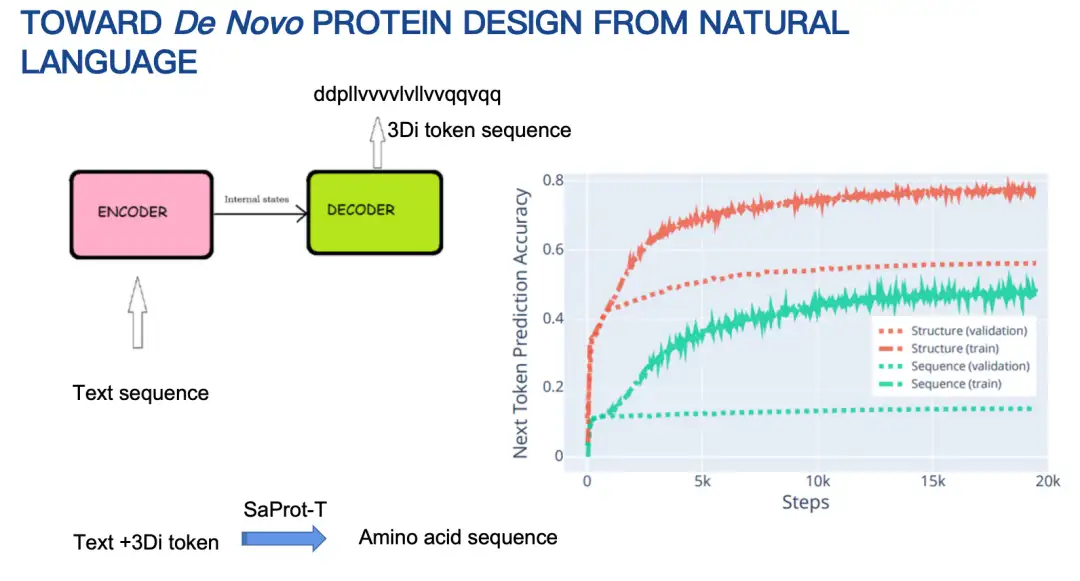
Let me briefly introduce the basic principles of Pinal (16 billion parameters).Initially, our idea was to use an encoder-decoder architecture, input text, and then output an amino acid sequence. However, after trying for a long time, the effect was not ideal. The main reason was that the space of amino acid sequences was too large, making prediction difficult.
Therefore, we adjusted our strategy and first designed the protein structure, and then designed the amino acid sequence based on the structure and textual hints. The protein structure here is also represented by discretized coding. The results show that the design method combined with the structure performs significantly better than the method of directly predicting the amino acid sequence in terms of Next Token Prediction Accuracy, as shown in the figure below.
We recently received a wet lab verification of Pinal from our collaborators.Pinal designed 6 protein sequences, 3 of which were expressed, and 2 sequences were verified to have corresponding enzyme catalytic activity. It is worth mentioning that in this work, we did not emphasize that we must design a protein that is better than the wild type. Our main goal is to verify whether the protein designed according to the text has the corresponding protein function.
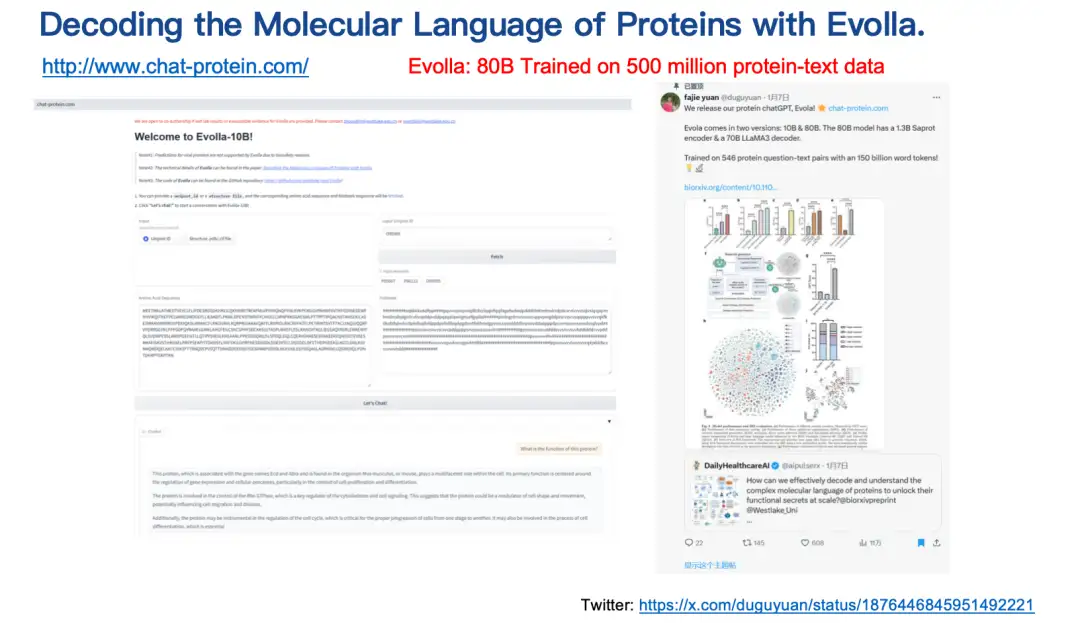
Evolla model: Decoding the molecular language of proteins
The last result introduced is the Evolla model.This is an 80 billion parameter protein language generation model, one of the largest open source biological models, designed to decode the molecular language of proteins.
By integrating protein sequence, structure and user query information,Evolla generates accurate insights into protein function.Users only need to enter the sequence and structure of the protein, then ask questions, such as introducing the basic function or catalytic activity of the protein, and simply click a button, and Evolla will generate a detailed description of about 200-500 words.
Evolla use address:
http://www.chat-protein.com/
Evolla paper address:
https://www.biorxiv.org/content/10.1101/2025.01.05.630192v2

It is worth mentioning that the training data and computing power of Evolla are very huge. Two of our doctoral students spent nearly a year just collecting and processing the training data. In the end, we generated more than 500 million high-quality protein-text pairs through synthetic data, covering hundreds of billions of word tokens. The model is quite accurate in predicting enzyme functions.But there are inevitably some illusion problems.
About the Team
Dr. Yuan Fajie of Westlake University is mainly engaged in applied scientific research related to traditional machine learning and interdisciplinary subjects, and focuses on the exploration of AI big models and computational biology. He has published more than 40 academic papers in top conferences and journals in the field of machine learning and artificial intelligence (such as NeurIPS, ICLR, SIGIR, WWW, TPAMI, Molecular Cell, etc.). For detailed information on team members and project contributors, please refer to the paper.
The research group has been conducting research in machine learning and AI + bioinformatics for a long time. You are welcome to apply for doctoral students, research assistants, postdoctoral fellows, and researcher positions in the research group. Students are welcome to visit the laboratory for internships. Those who are interested can send their resumes to [email protected].








