Command Palette
Search for a command to run...
Zhang Yang's Team at the National University of Singapore Developed a second-generation RNA Structure Prediction Algorithm That Surpassed SOTA in Multiple Benchmark Tests

Understanding the structure and function of RNA molecules has always been a core research direction in molecular biology and the pharmaceutical industry. RNA, especially non-coding RNA (ncRNA), can fold into specific structures and play an important role in a variety of cellular processes such as gene regulation (such as transcription and translation), catalysis, biological signal transduction, and stress response.
With the rapid development of high-throughput sequencing technology, RNA sequence data has grown exponentially, but the gap between known sequences and experimentally resolved RNA structures has continued to widen. Therefore, it has become increasingly urgent to resolve the atomic structure of RNA based solely on its original sequence. Researchers have developed a variety of RNA structure research methods, such as structural biology techniques such as X-ray crystallography, nuclear magnetic resonance spectroscopy, and cryo-electron microscopy (cryo-EM). Although these experimental techniques can provide higher resolution, the experimental resolution of RNA three-dimensional structure is often costly and, in some cases, difficult to achieve. Therefore,There is a growing demand for computational methods to predict high-quality RNA three-dimensional structure directly from sequence.
"Ab initio RNA structure prediction" refers to a method of directly predicting the three-dimensional structure of RNA from its sequence without relying on any experimental data or prior knowledge. The core of this method is to use computer simulation and computational chemistry technology to predict the three-dimensional conformation of RNA molecules through mathematical models and algorithms.
Recently, the latest research results from Professor Zhang Yang's team at the National University of Singapore have further promoted "Ab initio RNA structure prediction" to a higher level.Researchers proposed a high-precision RNA structure prediction framework based on deep learning, DRfold2.It integrates a pre-trained RNA composite language model (RCLM) and a denoising structure module for end-to-end RNA structure prediction. DRfold2 performs well in both global topology and secondary structure prediction compared to other state-of-the-art methods on multiple benchmarks.
Detailed analysis shows that this improvement mainly comes from RCLM's ability to capture co-evolutionary patterns and its efficient denoising process.This improves the unsupervised contact prediction accuracy of DRfold2 by more than 100% compared to existing methods.
The related results have been published on the preprint platform bioRxiv under the title "Ab initio RNA structure prediction with composite language model and denoised end-to-end learning".
Research highlights:
* DRfold2 integrates a pre-trained RNA composite language model (RCLM) and a denoising structure module for end-to-end RNA structure prediction
* Through a unique combination of composite language modeling, denoising-based end-to-end learning, and deep learning-guided post-optimization, DRfold2 opens up a new direction for "Ab initio RNA structure prediction"
* DRfold2 is highly complementary to AlphaFold3 and achieves statistically significant accuracy improvements after integration into the optimization framework

Paper address:
https://www.biorxiv.org/content/10.1101/2025.03.05.641632v1
Download DRfold2 RNA structure test dataset:
Dataset: Build an independent test dataset
In order to objectively evaluate the performance of DRfold2,The researchers constructed an independent test dataset containing 28 RNA structures.Their sequence lengths are all less than 400 nts and come from the following 3 categories:
* Latest RNA-Puzzles target sequences
* RNA target sequences in the CASP15 competition
* The most recently published RNA structures in the Protein Data Bank (PDB) database as of August 1, 2024
Notably, the researchers excluded large synthetic RNA structures from the CASP15 dataset because they deviate from RNA structures found in nature, which are the primary focus of functional analysis and drug design.
To ensure rigorous model evaluation, the training set only contains RNA structures published before 2024, and excludes RNAs with sequence similarity greater than 80% to the test dataset.
Download DRfold2 RNA structure test dataset:
Model architecture: a new RNA 3D structure prediction pipeline DRfold2
DRfold2 is a new RNA 3D structure prediction pipeline that consists of four core modules: (1) RNA Composite Language Model (RCLM), (2) RNA Transformer Block, (3) Denoising Structure Module, and (4) final model selection and optimization through the CSOR protocol, as shown in Figure A below:
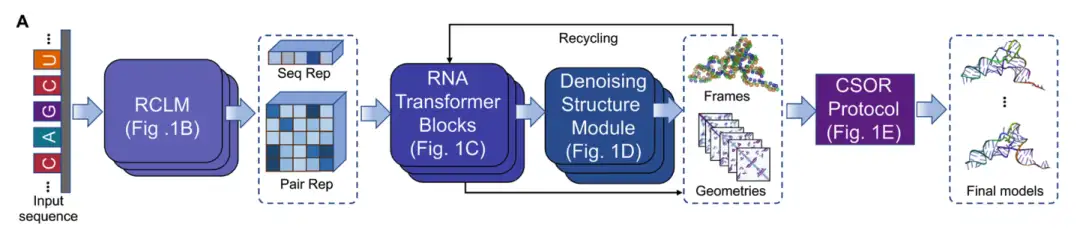
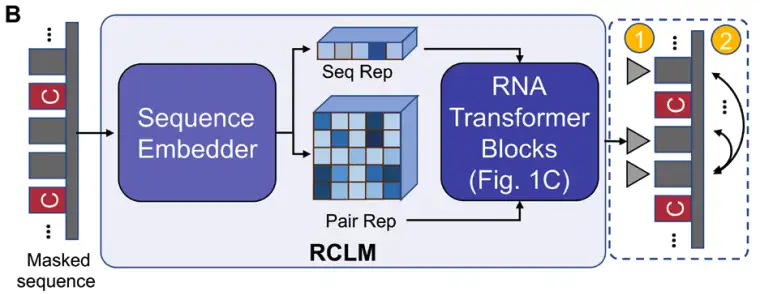
Starting with an input RNA sequence,DRfold2 first encodes the query sequence using a pre-trained RNA composite language model (RCLM).Generate sequence representation (Seq Rep) and pair representation (Pair Rep); RCLM is trained on large-scale unsupervised sequence data through the composite likelihood maximization method to achieve more efficient sequence pattern recognition, as shown in Figure B below:
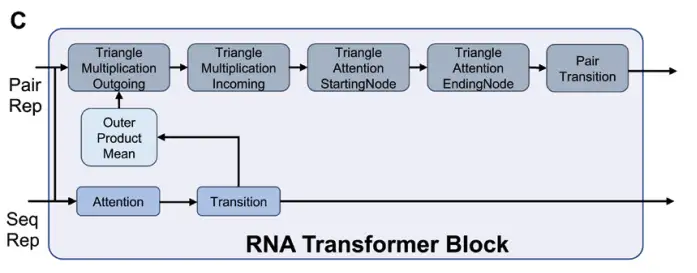
These sequences and pairwise representations are then fed into the RNA Transformer module for processing to generate key feature representations required for RNA structural folding, as shown in Figure C below:
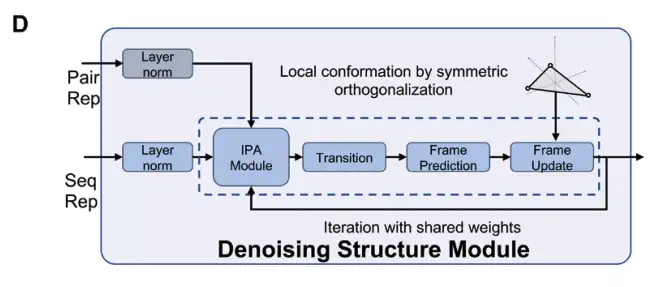
Next, DRfold2 uses the Denoising RNA Structure Module (DRSM) to generate RNA conformations in an end-to-end manner, as shown in Figure D below:
The final RNA structure model is screened and optimized through the post-processing CSOR protocol to select and refine the best model from the set of conformations generated at multiple checkpoints, as shown in Figure E below:
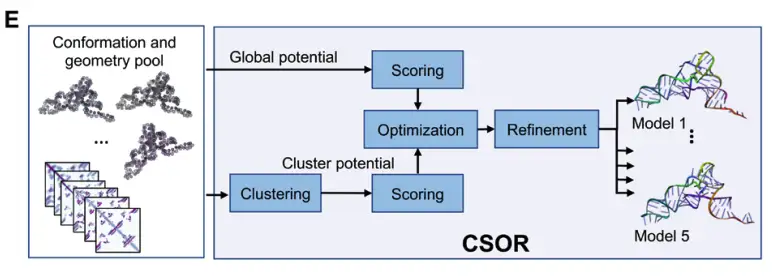
Although DRfold2 is named similarly to the team's earlier DRfold method, it introduces significant advances based on a completely different framework.The most important thing is the integration of a composite language model, which greatly enhances the ability of RNA sequence and pair representation.In addition, the prediction pipeline integrates a denoising RNA structure module (DRSM), which employs a controlled perturbation strategy to robustly learn structural transformations by efficiently correcting noisy RNA conformations.
The researchers have made the DRfold2 online server and local code publicly available at:
https://zhanglab.comp.nus.edu.sg/DRfold2
Research results: DRfold2 outperforms other state-of-the-art methods on multiple benchmarks
The researchers first compared DRfold2 with five state-of-the-art RNA structure prediction methods, including RNAComposer (fragment assembly and optimization based), trRosettaRNA (deep learning method), RhoFold (end-to-end deep learning method), RoseTTAFoldNA (end-to-end deep learning method) and DeepFoldRNA (deep learning method).
As shown in the figure below, the researchers compared the TM-score and RMSD evaluation results of DRfold2 and the benchmark method at different sequence similarity thresholds (50%-80%). Among them, TM-score is a length-independent scoring function used to evaluate the overall quality of the predicted RNA structure, with a value range of 0-1. The higher the value, the higher the similarity between the predicted structure and the true structure.
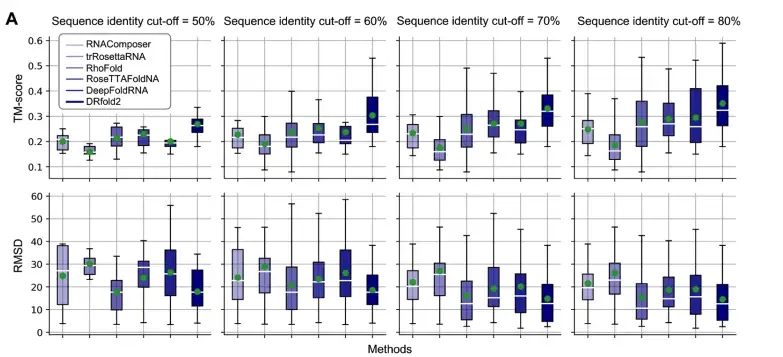
The results show that DRfold2 always obtains the highest average TM-score under all sequence similarity thresholds.For example:
* Under the 80% similarity threshold, the average TM-score of DRfold2 is 0.351, which is 18.6% higher than the second-ranked DeepFoldRNA (TM-score=0.296).
* Under the 50% similarity threshold (the most stringent test set), DRfold2 can still obtain an average TM-score of 0.269, which is 17.5% higher than the second-ranked RoseTTAFoldNA (TM-score=0.229).
* In addition, the RMSD (root mean square deviation) of DRfold2 at all sequence similarity thresholds is always lower than that of all control methods, indicating that its predicted structure is closer to the real RNA structure.
The researchers further used the chimpanzee CPEB3 HDV-like ribozyme (PDB ID: 7QR3) as an example. The RNA is 69 nucleotides long and analyzed the prediction effects of different methods on its RNA tertiary structure. The results are as follows:
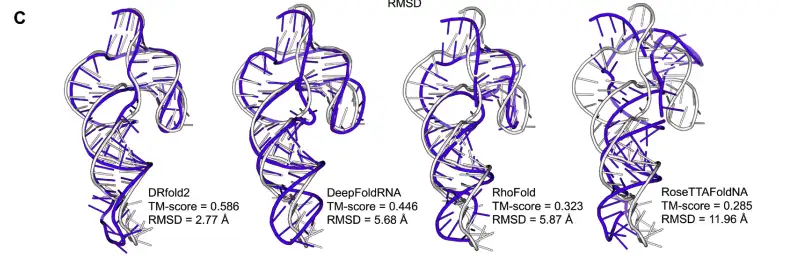
* DRfold2 accurately captured the overall topological structure of the ribozyme, with a TM-score of 0.586 and a RMSD of only 2.77 Å.
* DeepFoldRNA performs well in terms of overall helical arrangement, but the direction of the hairpin loop deviates significantly, resulting in an RMSD as high as 5.68 Å, which is twice the deviation of DRfold2.
* RhoFold and RoseTTAFoldNA have larger errors in spatial prediction in junction regions, causing the TM-score to drop to 0.323 and 0.285.
* The highest sequence similarity between the target RNA and the training dataset is only 60.9%, indicating that DRfold2 can still provide reliable structure predictions for new RNA sequences in the absence of homologous templates.
These results show that:The comprehensive probabilistic representation provided by higher-order language models like RCLM significantly enhances the ability to learn co-evolving patterns and spatial constraints.Thus, more accurate 3D RNA structure modeling was achieved through the end-to-end network of DRfold2.
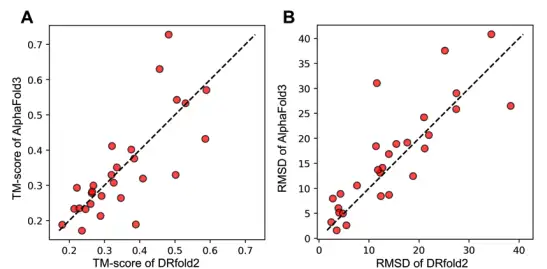
On this basis, in order to compare the performance of DRfold2 and AlphaFold3 in RNA 3D structure prediction, the researchers also submitted the RNA sequences in the test set to the AlphaFold server and used the default seed configuration to obtain the predicted structure of AlphaFold3.The average TM-score (0.351) and RMSD (14.6 Å) of DRfold2 are slightly higher than those of AlphaFold3 (0.345 and 16.0 Å).
What’s more worth mentioning is that although DRfold2 and AlphaFold3 show similar overall performance, the results in the figure below highlight the strong complementarity between the two, especially when the prediction deviates significantly from the diagonal line——By incorporating AlphaFold3’s predictions as an additional potential function term into the DRfold2 optimization framework, the researchers achieved statistically significant improvements in both TM-score and RMSD.
Professor Zhang Yang's team has been focusing on AI and computational biology research for many years
The DRfold2 proposed in this study is actually an upgraded version of the DRfold model previously proposed by Professor Zhang Yang's team.
In September 2023, Professor Zhang Yang's team published a paper titled "Integrating end-to-end learning with deep geometrical potentials for ab initio RNA structure prediction" in the journal Nature Communications.
This study reports a new technology, DRfold, for accurately predicting the three-dimensional structure of RNA.The core innovation lies in the introduction of two complementary potential energy functions: FAPE potential and geometric potential.They are trained through two independent Transformer networks, and together they form a deep learning potential for RNA structure prediction. The calculation results show that compared with previous RNA structure computer prediction methods, DRfold surpasses these methods in multiple performance indicators.

Paper address:
https://www.nature.com/articles/s41467-023-41303-9
From DRfold to DRfold2, Professor Zhang Yang's team has continued to focus on artificial intelligence and computational biology research for many years. His laboratory is one of the earliest laboratories to carry out protein and RNA structure prediction research based on deep machine learning. He has won honors such as the Sloan Award, the National Science Foundation Career Award, and the University of Michigan Basic Science Research Award. Since 2015, he has been selected into the Thomson Reuters/Clarivate Analytics Global Highly Cited Scientists List 7 times. The I-TASSER algorithm (https://zhanggroup.org/I-TASSER/), Since 2006, it has been rated as the most accurate automated protein structure prediction method in the worldwide CASP experiment for nine consecutive times.
On January 2, 2024, Professor Zhang Yang's team published a paper titled "Improving deep learning protein monomer and complex structure prediction using DeepMSA2 with huge metagenomics data" in the journal Nature Methods.
The study developed two new software to improve the accuracy of protein interaction structure prediction. The authors developed DeepMSA2, which uses recursive dynamic programming and hidden Markov model algorithms to quickly extract high-quality MSA data from massive metagenomic sequence libraries, and then uses the newly developed DMFold software to construct the three-dimensional structure of protein complexes.
Experimental results show that DMFold/DeepMSA2 is significantly better than AlphaFold2 and other algorithms in predicting the structure of protein complexes.https://zhanggroup.org/DMFold) algorithm won the championship in protein complex structure prediction in the latest protein structure prediction competition (CASP15).

Paper address:
https://www.nature.com/articles/s41592-023-02130-4
Recently, the team has further expanded its research direction to include RNA and short peptide design and structure prediction, and explore topics related to drug design. In the future, I believe that Professor Zhang Yang will continue to lead his team to explore the mysteries of biology.
References:
1.https://www.biorxiv.org/content/10.1101/2025.03.05.641632v1
2.https://mp.weixin.qq.com/s/X_VJ-WOWEP08p5GAJOgq9A








