Command Palette
Search for a command to run...
Selected for AAAI 2025! Tsinghua University/UCL Pioneered the Protein-rna Language Model Fusion Solution, Combining Affinity Prediction to Refresh SOTA
Alzheimer's disease, Parkinson's disease, epilepsy... These "notoriously terrifying" neurodegenerative diseases are invisible killers of the health of the elderly, and the occurrence of these diseases is often related to the abnormal binding between protein and RNA.
In the biomedical field, studying protein-RNA binding is of vital importance because it plays a central role in multiple biological processes such as gene expression regulation, RNA processing and splicing, translation regulation, and cellular stress response.Understanding the mechanism of protein-RNA binding is the key to revealing complex gene regulation processes and analyzing the genetic basis of diseases. At the same time, protein-RNA interactions also have important applications in RNA targeted therapy, providing new directions for the treatment of cancer, genetic diseases and viral diseases.
Recently, among the selected achievements announced at the 39th Annual AAAI Conference on Artificial Intelligence (AAAI 2025), the top international artificial intelligence conference,A joint team from Tsinghua University, University College London, Monash University, and Beijing University of Posts and TelecommunicationsproposeThe CoPRA model has attracted wide attention in the industry and was selected for the Oral segment.
This is the first attempt to combine the protein language model (PLM) and RNA language model (RLM) through a complex structural architecture for protein-RNA binding affinity prediction.To test the performance of CoPRA, the researchers compiled the largest protein-RNA binding affinity dataset from multiple data sources and evaluated the model performance on 3 datasets. The results showed that CoPRA achieved state-of-the-art performance on multiple datasets.
The related results are titled "CoPRA: Bridging Cross-domain Pretrained Sequence Models with Complex Structures for Protein-RNA Binding Affinity Prediction" and have been published as a preprint on arXiv.

Paper address:
https://arxiv.org/abs/2409.03773
CoPRA Warehouse Address:
https://github.com/hanrthu/CoPRA
The open source project "awesome-ai4s" brings together more than 200 AI4S paper interpretations and provides massive data sets and tools:
https://github.com/hyperai/awesome-ai4s
Biomedical research continues to advance protein-RNA interactions
Over the past years, researchers in the biomedical field have never stopped studying protein-RNA interactions and have made considerable progress.
As one of the most important techniques for RNA research, CLIP experimental technology can analyze the binding map of RNA binding protein (RBP) on the entire transcriptome, which is the basis for systematically understanding the function of an RBP and its regulatory mechanism. However, CLIP experiments are time-consuming and labor-intensive, and can only provide the RNA binding site of a certain RBP in a specific cell environment at a time, and have high requirements for experimental materials. However, the binding of protein and RNA may change greatly with changes in the cell environment, but studying the regulation of protein on RNA requires binding information in the same cell environment.
In order to solve the problem of dynamic changes in RBP binding in different cellular environments,In February 2021, Zhang Qiangfeng's research group at Tsinghua University's Advanced Innovation Center for Structural Biology published a research result titled "Predicting dynamic cellular protein-RNA interactions by deep learning using in vivo RNA structures" in the journal Cell Research. This work used the icSHAPE experiment to analyze the RNA secondary structure maps of seven common cell types, and developed an artificial intelligence algorithm to integrate the experimentally obtained intracellular RNA structure and the RBP binding information of the corresponding cellular environment, and established a new method PrismNet for predicting the dynamic binding of intracellular RBP based on intracellular RNA structure information.
In order to predict protein-RNA binding affinity, several computational methods have been proposed in the industry.Includes sequence-based and structure-based methods. Sequence-based methods use different sequence encoders to process protein and RNA sequences, respectively, and subsequently model the interactions between them. However, since the binding affinity is mainly determined by the structure of the binding interface, the performance of these methods is often limited. Other recently proposed methods focus on extracting structural features of the binding interface, such as energy and contact distance. Based on these extracted features, researchers have developed structure-based machine learning methods that can be used for affinity prediction. However, due to the limitations of the dataset size, these methods have limited generalization capabilities on new samples and are highly dependent on feature engineering.
With the rise of artificial intelligence technology, many protein language models (PLMs) and RNA language models (RLMs) have been developed, which have demonstrated excellent performance and generalization capabilities in various downstream tasks.At the same time, since the three-dimensional structure of proteins/RNAs is crucial to understanding their functions, incorporating structural information into language models has also become a new trend.
For example, a team composed of the University of Missouri, the University of Kentucky and the University of Alabama used multi-view contrastive learning technology to incorporate key protein structure information into the protein language model. Based on this concept, the team developed S-PLM: a protein language model with the ability to perceive protein 3D structural information. S-PLM has demonstrated excellent performance in multiple protein prediction tasks. After training with lightweight tuning tools, S-PLM's performance in tasks such as protein function prediction, enzyme reaction category prediction and secondary structure prediction reaches or exceeds the current most advanced methods. The related research was published on bioRxiv under the title "S-PLM: Structure-aware Protein Language Model via Contrastive Learning between Sequence and Structure".
However, although current industry research has demonstrated the great potential of biological language models driven by structural information in interactive tasks, work combining pre-trained models from different biological fields is still rare.In CoPRA, jointly proposed by Tsinghua University, University College London, Monash University, and Beijing University of Posts and Telecommunications, an attempt was made for the first time to combine protein and RNA language models with complex structural information for the prediction of protein-RNA binding affinity.
Designing a lightweight Co-Former model to build CoPRA
Overall, the construction process of the CoPRA model is shown in the following figure:
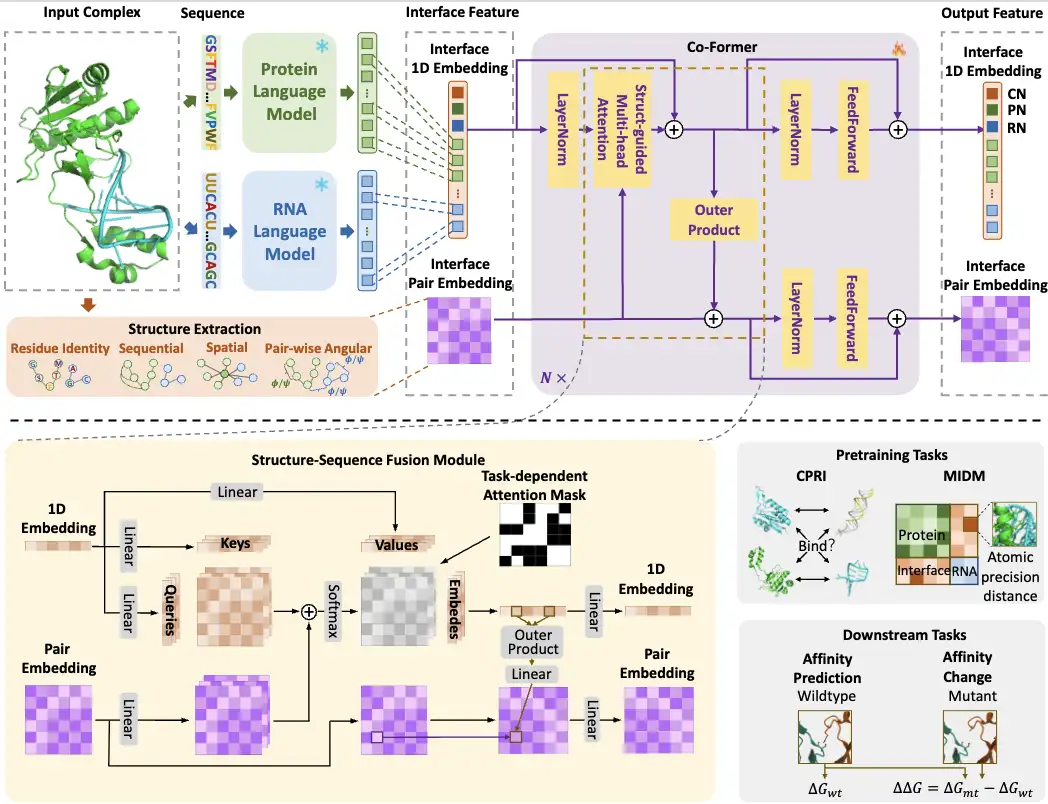
first,The researchers input protein and RNA sequences into PLM and RLM respectively, and then select the embedding at the interaction interface from the output of the two language models as the sequence embedding for subsequent cross-modal learning. At the same time, they also extract structural information (interface feature) from the interaction interface as paired embedding.
Then,The researchers designed a lightweight Co-Former model that combines interface sequence embeddings from two language models with complex structural information to form a structure-sequence fusion module. Specifically, Co-Former fuses 1D and paired embeddings through structure-guided multi-head self-attention and outer product modules, and applies task-related attention masks. The output special nodes and paired embeddings of Co-Former are used according to different tasks, including two pretraining tasks and two downstream affinity tasks.
The researchers also proposed a dual-range pre-training strategy for Co-Former.To model coarse-grained contrastive interaction classification (CPRI) and fine-grained interface distance prediction (MIDM), learned with atomic-level accuracy.
To evaluate the performance of CoPRA and other models,The researchers needed to solve the problem of the lack of unified annotation standard datasets. Therefore, they collected samples from three public datasets: PDBbind, PRBABv2, and ProNAB, compiled the largest protein-RNA binding affinity dataset PRA310, and evaluated their model's ability to predict protein-RNA binding affinity on the PRA310 and PRA201 datasets.
*PRA201 dataset: a subset of PRA310, each complex contains only one protein chain and one RNA chain, and has stricter length constraints
CoPRA performs best in predicting protein-RNA binding affinity
As shown in the table below, the de novo trained version of CoPRA achieves the best performance on the PRA310 dataset. In addition, most methods that use LM embeddings as input outperform other methods, indicating the great potential of combining pre-trained unimodal LMs for affinity prediction.
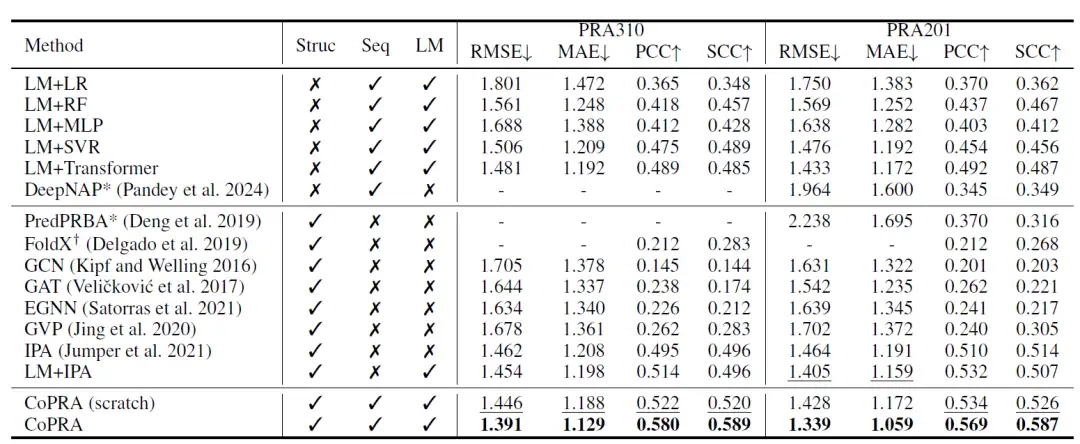
Subsequently, the researchers pre-trained the model using their collated unsupervised dataset PRI30k, significantly improving its overall performance on both datasets. On the PRA310 dataset, CoPRA achieved an RMSE of 1.391, MAE of 1.129, PCC of 0.580, and SCC of 0.589, far outperforming the second-best model CoPRA (de novo training version). PredPRBA and DeepNAP support protein-RNA affinity prediction, and the researchers compared the performance of these methods on the PRA201 dataset, showing that their performance on PRA201 was significantly lower than their reported results, despite at least 100 samples in PRA201 appearing in their training sets, indicating that these methods have poor generalization capabilities.
CoPRA is stronger in predicting the effect of mutations on binding affinity and has excellent generalization ability
To further evaluate the model's fine-grained understanding of affinity, the researchers redirected the model to predict the effects of single-point mutations in proteins on protein-RNA complexes. Referring to related studies on the prediction of protein mutation effects, the researchers averaged the metrics at the level of each complex and evaluated the zero-shot performance and fine-tuning performance of CoPRA after pre-training on PRI30k and tuning on PRA310.
As shown in the table below, after fine-tuning using the cross-validation set of mCSM, the model proposed in this study surpassed other models in all four indicators, with RMSE of 0.957, MAE of 0.833, PCC of 0.550, and SCC of 0.570.
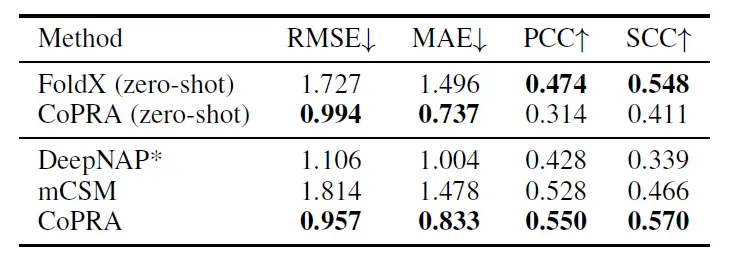
This superior performance stems from the dual pre-training objective despite not seeing any mutant complex structures, demonstrating the generalization ability of CoPRA on different affinity-related tasks.
Breakthrough progress in multimodal protein language models
The essence of the research idea introduced above is to combine multiple biological modalities such as proteins and RNA with complex structural information, which is the so-called multimodal learning. Simply put, multimodal learning is to integrate various types of data into one model for modeling under the framework of deep learning.
In the past few years, with the rapid development of big language models, researchers have begun to try to apply them to the field of protein science to accurately understand and predict the function, structure and properties of proteins. However, previous protein-oriented big language models mainly process amino acid sequences as text, failing to fully utilize the rich structural information of proteins.Today, progress in multimodal learning has provided new ideas for more and more related research.
For example, in the field of drug development, accurate and effective prediction of the binding affinity between proteins and ligands is crucial for drug screening and optimization. However, previous studies have not considered the important role of molecular surface information in protein-ligand interactions.Researchers from Xiamen University proposed a novel multimodal feature extraction (MFE) framework.This framework combines information from protein surface, 3D structure, and sequence for the first time, and uses a cross-attention mechanism to align features between different modalities. Experimental results show that this method achieves state-of-the-art performance in predicting protein-ligand binding affinity. The related research was published in Bioinformatics in June 2024 under the title of "Surface-based multimodal protein–ligand binding affinity prediction".
In December 2024, a research team from East China Normal University and other institutions proposed an innovative solution, EvoLLama.This is a framework that integrates protein structure encoders, sequence encoders, and large language models for multimodal fusion. In the zero-shot setting, EvoLLama demonstrates strong generalization capabilities, improving the performance of other fine-tuned baseline models by 1%-8%, and surpassing the current most advanced supervised fine-tuning model with an average performance of 6%. The relevant research results have been published as a preprint on arXiv under the title "EvoLlama: Enhancing LLMs' Understanding of Proteins via Multimodal Structure and Sequence Representations".
Of course, multimodal learning is just one of the research options available. In the future, by using more machine learning methods to study the surface of proteins, biologists can have a deeper understanding of how they interact with other biological molecules, thereby providing assistance for the development of new drugs.
References:
1.https://arxiv.org/abs/2409.03773
2.https://www.frcbs.tsinghua.edu.cn/index.php?c=show&id=873
3.https://www.sohu.com/a/846589543_121124715








