Command Palette
Search for a command to run...
The Success Rate Can Reach 100%. Drug Development Company Cellarity Teamed up With NVIDIA to Optimize Targeted Molecules Based on Reinforcement Learning

From ancient times to the present, mankind has never stopped fighting against diseases. The emergence of a new drug may save thousands of lives and even extend the overall human lifespan.
Looking back at the history of drug development over the past century, there are many interesting stories. For example, in the early 19th century, a German pharmacist's assistant, Zertina, soaked opium in hot water and extracted it with ammonia water to separate a pile of white powder from the opium. He fed this white powder to a dog, and the dog soon fainted after eating it.So he named it morphine after the Greek god of dreams, Morpheus.Therefore, morphine is generally considered to be the world's first active ingredient isolated from a plant, and is also considered the starting point of modern drug innovation.
Subsequently, pharmacists gradually mastered the technology of synthesizing chemical drugs, and German pharmacist Selmann synthesized acetylsalicylic acid, the predecessor of aspirin. At the beginning of the 20th century,Companies' demand for new drugs has driven the development of high-throughput screening technology, which enables scientists to screen and test large numbers of compounds with greater efficiency. At the beginning of the 21st century,Researchers have begun to explore more precise and effective drug treatments, among which targeted drugs have become a hot research direction.
Today, the rapid development of artificial intelligence technology has brought new possibilities to drug discovery. AI can help pharmacists verify drug targets and optimize drug structure design more quickly, and even directly generate molecules with specific physical and chemical properties or biological activities, greatly accelerating drug discovery.
In this context,Researchers from the life science company Cellarity and NVIDIA have jointly proposed a novel targeted molecular optimization method based on latent reinforcement learning, MOLRL.The method combines a powerful generative model pre-trained on a large number of chemical datasets with a state-of-the-art reinforcement learning (RL) algorithm for continuous space optimization. The researchers applied the method to drug discovery-related tasks, using common benchmarks and comparing with state-of-the-art methods, and found that MOLRL showed superior or competitive performance in a variety of tasks, especially in targeted molecule generation and multi-parameter optimization.
The related results were published on ChemRxiv under the title "Targeted Molecular Generation With Latent Reinforcement Learning".
Paper address:
Follow the official account and reply "targeted molecule optimization" to get the complete PDF
The open source project "awesome-ai4s" brings together more than 100 AI4S paper interpretations and provides massive data sets and tools:
https://github.com/hyperai/awesome-ai4s
Route selection: directly modifying molecules vs. operating in latent space
Drug development is a very complex process - in addition to biological activity, compounds should also have multiple other properties to be selected as clinical candidate drugs. And those compounds identified as having therapeutic activity, usually called "candidate compounds", are not fixed in structure, but will be modified in a long iterative cycle to solve problems such as insufficient solubility and insufficient activity.
In an iterative process, pharmacists usually transform the initial molecule to design analogs based on their intuition or through reaction-based library enumeration. However, given the large size of chemical space, design becomes extremely difficult even for a single molecule, requiring an exhaustive evaluation of the entire chemical space. Computational methods for targeted molecule generation can efficiently explore chemical space and recommend structures that have not been explored before to chemists.
Currently, methods for target molecule generation and optimization can be divided into two categories:The first method is to operate directly on the molecular structure.to identify structural modifications that improve target properties;The second category of methods operates in the latent space of the generative model.Modify the molecular structure indirectly through its latent representation.
Method 1 can perform structural modifications by inserting or deleting atoms or chemical bonds, and the industry has made considerable progress.
It was reported that in November last year, a team led by Professor Yoonsu Park of the Korea Advanced Institute of Science and Technology (KAIST) developed an innovative single-atom editing technology. This technology introduces photocatalysts toSingle-atom editing of drug molecules was successfully achieved at room temperature and pressure.The "molecular scissors" technology developed by the team can accurately cut and connect five-membered ring structures, replacing oxygen atoms with nitrogen atoms, changing the molecular properties and improving the efficacy of drugs. The relevant research results were published in Science under the title "Photocatalytic furan-to-pyrrole conversion".

However, it is not easy to perform surgery on molecules at will. On the one hand, structural modifications may violate chemical rules, resulting in invalid molecular structures. On the other hand, since molecular structures are discrete in nature, and adding or removing chemical bonds involves discrete operations, this discreteness will lead to discontinuous gradients in the optimization process, making it difficult to effectively apply gradient-based methods.
Compared with method 1,The second approach transforms the optimization task into a continuous optimization problem, exploits the latent space of the generative model, and adopts continuous space optimization algorithms such as gradient descent.Despite this, chemical validity remains a challenge as there is no guarantee that a point in the latent space corresponds to a valid molecule. However, by using novel architectures as well as training modifications, generative models have made significant progress in improving validity and continuity of the latent space.
In the research of Cellarity and NVIDIA, the researchers proposed MOLRL to optimize in the latent space of the pre-trained generative model by using the proximal policy optimization (PPO) method.
MOLRL, a targeted molecular optimization method based on latent reinforcement learning
How does the MOLRL framework work?
The MOLRL framework consists of two parts: a latent space generative model and a reinforcement learning (RL) agent.
The generative model is a pre-trained encoder-decoder model whose latent space encodes the chemical space in which the RL agent operates. The RL agent is trained using the PPO method.To navigate in the latent space; The reward function provides feedback to the agent,Help them learn how to navigate the space,Identify molecules with desired properties.
As shown below: The latent representation "z" of the input molecule is perturbed by the action "a" extracted from the policy network output. The perturbed latent vector "z'" is decoded into molecules and scored by the reward function. The state "z", action "a" and reward "R" are collected to update the policy network.
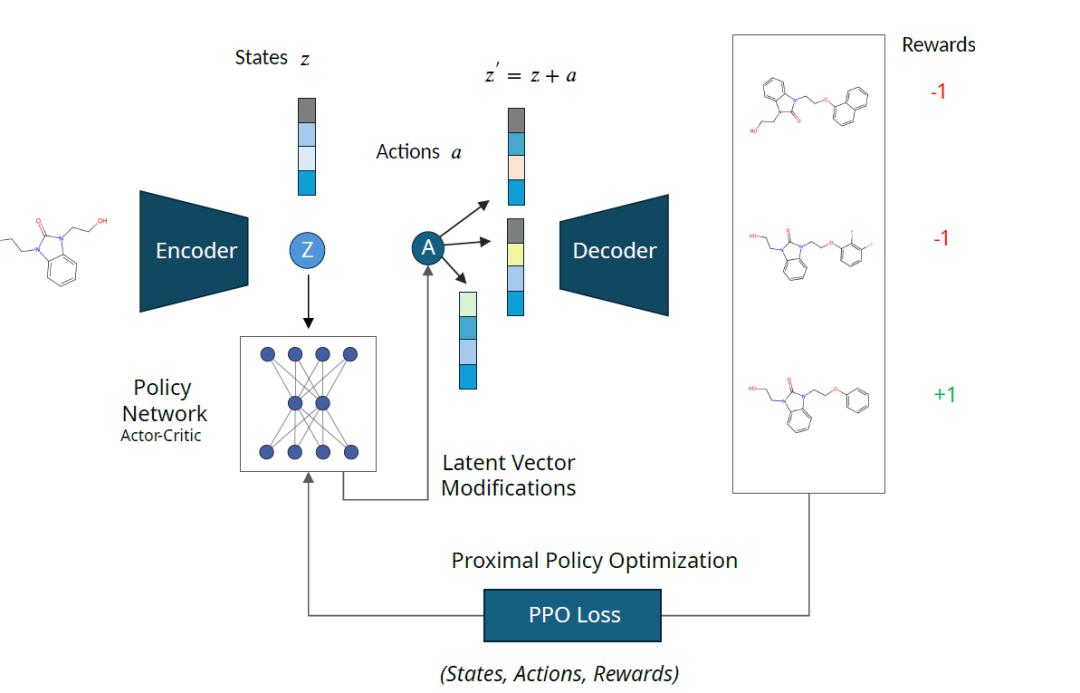
The framework is independent of the architecture of the encoder and decoder, however, the characteristics of the latent space will greatly affect the optimization performance. Therefore, the researchers evaluated the performance of MOLRL on two different encoder-decoder architectures, namely variational autoencoder (VAE) and autoencoder trained based on mutual information machine learning (MolMIM).
The reinforcement learning (RL) agent is responsible for navigating the latent space to identify molecules with the desired molecular properties. The researchers used PPO, or the proximal policy optimization algorithm, to train the RL agent.The PPO algorithm guides the agent to find an optimal path in the latent space by optimizing the policy to maximize the long-term cumulative reward.The reward function is the core of the MOLRL framework, which provides feedback to the agent based on the target properties of the molecule (such as drug-likeness, synthetic accessibility, target binding, etc.).
How does the MOLRL framework perform?
To evaluate the performance of the MOLRL framework, the researchers designed a multi-objective optimization task and compared it with the current state-of-the-art optimization methods.
Specifically, the researchers applied MOLRL to generate biologically active molecules, targeting two targets, while optimizing drug-likeness (QED) and synthetic accessibility (SA). The selected biological targets were two kinases associated with Alzheimer's disease, GSK3β and JNK3. According to the evaluation strategy of Jin et al., the researchers recorded the top 5,000 molecules with the highest reward values generated during the optimization process and calculated the following three indicators: success rate; novelty; diversity.
The following table shows the performance of MOLRL trained in the VAE-CYC latent space and MOLRL trained in the MolMIM space, as well as the performance comparison of current state-of-the-art molecular optimization methods reported in the literature.
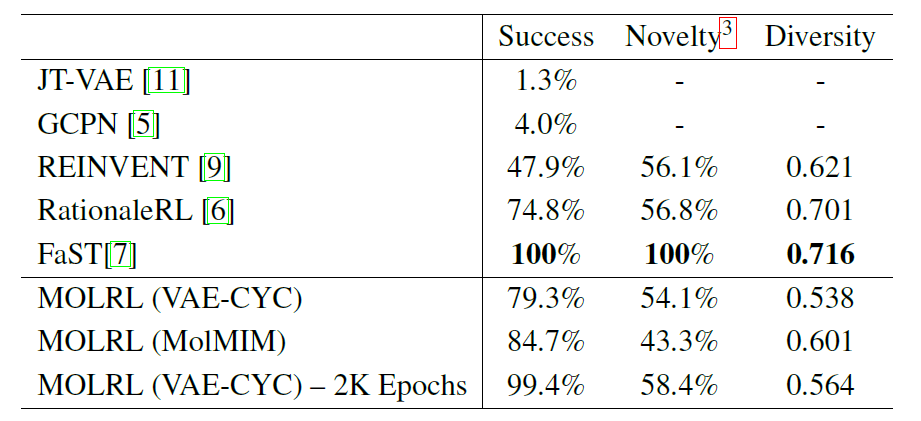
As shown in the table, FaST constructs molecular graphs by combining molecular fragments using reinforcement learning (RL).It shows higher success rate among all compared methods. FaST and RationaleRL have advantages in terms of diversity and novelty, and both methods exploit prior knowledge. REINVENT and MOLRL both start from random molecules that may be far from the training range of ML classifiers.MOLRL still achieves comparable novelty to RationaleRL and achieves the highest success rate.
Using prior knowledge as a starting point can bring certain advantages, but it can also limit novelty and the ability of the algorithm to discover new skeletons. In addition, the applicability of such methods is limited when no prior knowledge is available, such as when studying unexplored targets.
In addition to multi-objective optimization tasks, a common practice in drug discovery is to identify a chemical skeleton that is known to bind to a certain target or target class and use it as a starting point for chemical design and optimization. Therefore, the paper further verifies the ability of MOLRL to optimize multi-objective properties while retaining the specified molecular skeleton. As shown in the following table,When optimizing molecules containing an aminopyrimidine backbone, MOLRL achieved a success rate of 100%.
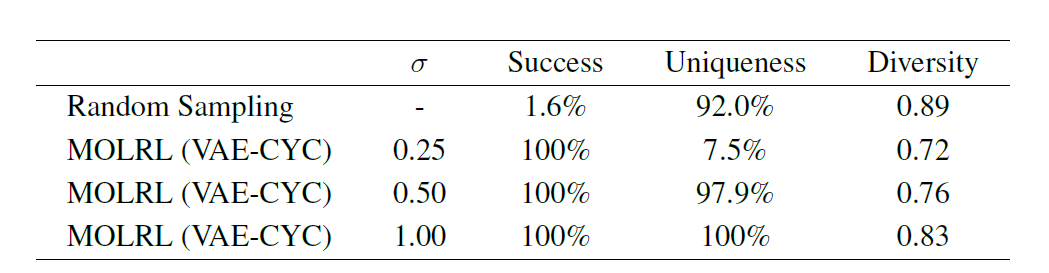
In summary, MOLRL shows superior or competitive performance in a variety of tasks compared to existing methods.Especially in targeted molecule generation and multi-parameter optimization.
AI is a key step in improving drug discovery efficiency
How much resources does it take to develop a new drug? The pharmaceutical industry has a famous "double ten rule", which means that it takes 10 years and $1 billion for a new drug to be discovered and marketed. According to a recent report released by Deloitte, if the cost of failed clinical trials is taken into account, the average cost for the world's top pharmaceutical companies to successfully bring a new drug to market isIt has increased from US$1.188 billion in 2010 to US$2.284 billion in 2022.
A key step in drug discovery is to find a batch of candidate molecules for computational research or synthesis and characterization, which is a difficult task because the chemical space of potential molecules is huge and requires extremely high trial and error costs. Today, artificial intelligence and machine learning can effectively improve the efficiency of this step.
October 31, 2023Novartis Institutes for BioMedical Research and Microsoft Research Center for Scientific Intelligence collaborated toA research paper titled “Extracting medicinal chemistry intuition via preference machine learning” was published in Nature Communications.
The researchers asked 35 medicinal chemists to choose their preferred molecule from 5,000 pairs of molecules, and then used their answers to make a ranking game to train a machine learning model, which was then asked to score the molecules. This score is basically unaffected by other properties that have been used as features in the field before, because it comes from years of knowledge accumulation in the industry.
The model can partially reproduce the collective knowledge accumulated by professional chemists in their work, which is often called "chemistry intuition", making future drug development more efficient.
In March 2024, Insilico Medicine, a leading AI pharmaceutical company, published a scientific research paper in Nature Biotechnology, detailing the use of an artificial intelligence platform to discover the novel target TNIK for the treatment of IPF, and the subsequent process of using a generative chemistry platform to design the ISM001-055 molecule.
ISM001-055 is a world-first small molecule inhibitor.Targeting TNIK (Traf2/NCK interacting kinase) for the treatment of idiopathic pulmonary fibrosis (IPF). Insilico Medicine said that generative AI can greatly improve R&D efficiency, reduce R&D costs, and increase R&D success rate in the early stages of R&D. Taking the molecules against idiopathic pulmonary fibrosis as an example, from early target discovery to the determination of preclinical candidate compounds,It took only 18 months and invested $2.6 million in research and development.
According to a research report from Fortune Business Insights, the global market size of artificial intelligence in drug discovery is $3 billion in 2022, and is expected to grow from $3.54 billion in 2023 to $7.94 billion in 2030, with a compound annual growth rate of 12.21%. In the future, AI technology has great potential to promote changes in the pharmaceutical industry.
References:
1.https://mp.weixin.qq.com/s/OL7TJQcUE-ubhUDyc7GBzQ
2.https://www.thepaper.cn/newsDetail_forward_29097303
3.https://news.bioon.com/article/6127e7234091.html
4.https://bydrug.pharmcube.com/news/detail/49720140c1e9d57ac3c7cfe20ef7f8be
5.https://mp.weixin.qq.com/s/UGAXWMhPlSg2hFnI5ghr1w









