Command Palette
Search for a command to run...
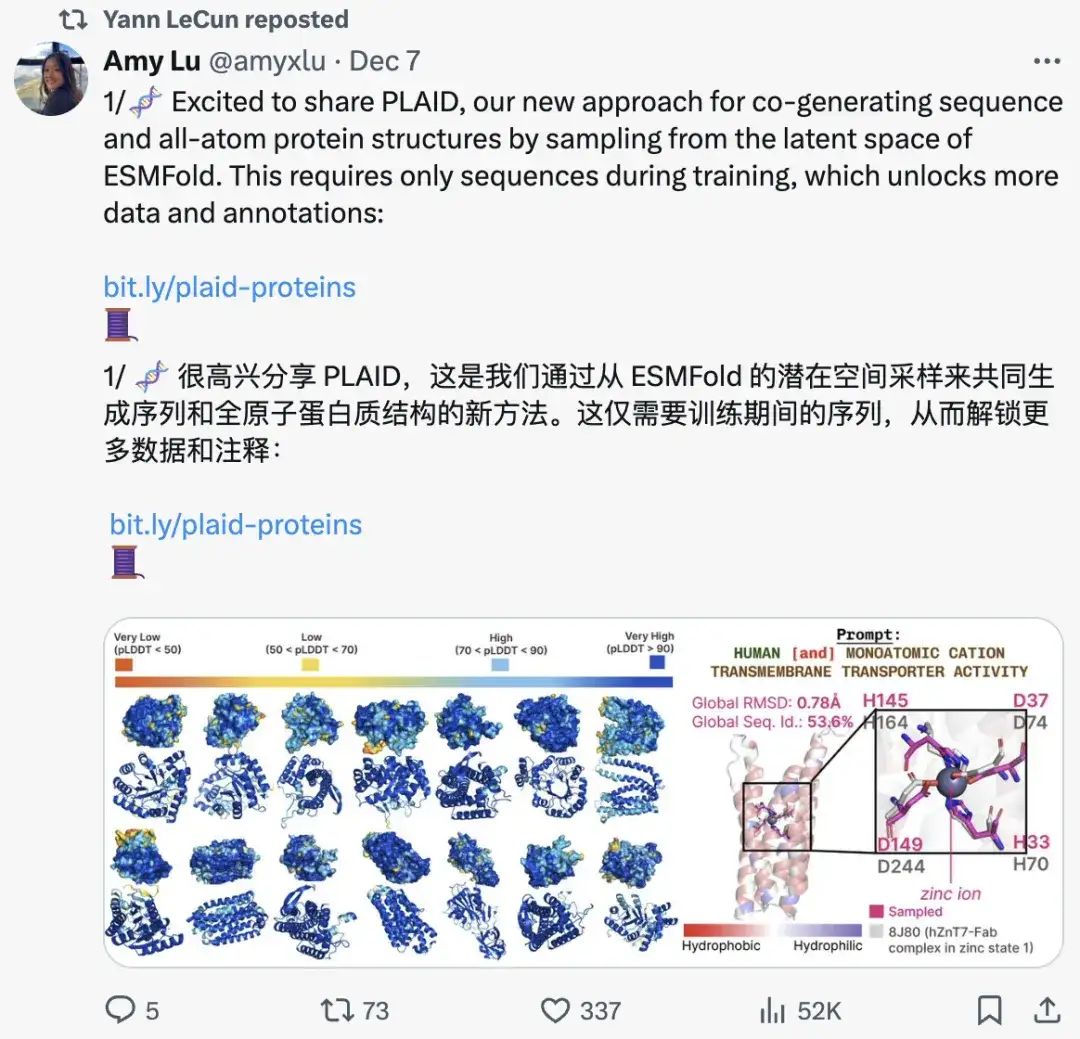
LeCun Forwarded, UC Berkeley Et al. Proposed a Multimodal Protein Generation Method PLAID, Which Generates Sequences and all-atom Protein Structures at the Same Time

Over the past years, scientists have continued to explore the structure and composition of proteins in order to better unravel the "code of life."Protein function is determined by its structure, including the identity and position of side-chain and main-chain atoms and their biophysical properties, which are collectively referred to as the all-atomic structure.However, to determine the placement of side chain atoms, the sequence must be known first. Therefore, all-atom structure generation can be viewed as a multimodal problem that requires the generation of both sequence and structure.
However, existing protein structure and sequence generation methods usually treat sequence and structure as independent modes. Structure generation methods usually only generate main chain atoms. Methods targeting all-atom design usually require the use of external models to alternate between structure prediction and anti-folding steps, etc.
To address these challenges, a research team from the University of California, Berkeley (UC Berkeley), Microsoft Research, and Genentech proposed a multimodal protein generation method called PLAID (Protein Latent Induced Diffusion), which can achieve multimodal generation by mapping from richer data modalities (such as sequences) to scarcer modalities (such as crystal structures).To validate the approach, the researchers conducted experiments on 2,219 functions from the Gene Ontology and 3,617 organisms across the tree of life.Even though no structural input is used during training, the generated samples show strong structural quality and consistency.
The related research is titled "Generating All-Atom Protein Structure from Sequence-Only Training Data" and has been submitted to the top conference ICLR 2025. "AI Godfather" Yang Likun also reposted this achievement on the social platform.
PLAID project open source address:
http://github.com/amyxlu/plaid
Research highlights:
* Focusing on the large protein language model ESMFold and all-atom structure generation, the researchers proposed a controllable diffusion model that can simultaneously generate sequences and all-atom protein structures, requiring only sequence input during training.
* The approach exploits structural information encoded in pre-trained weights rather than training data and increases the availability of sequence annotations for controllable generation.
* Although the ESMFold model is used in the paper, the method can be applied to any forecasting model.
Paper address:
https://www.biorxiv.org/content/10.1101/2024.12.02.626353v1
The open source project "awesome-ai4s" brings together more than 100 AI4S paper interpretations and provides massive data sets and tools:
https://github.com/hyperai/awesome-ai4s
Research highlights at a glance
Dataset
The researchers used the September 2023 release of the Pfam database, which contains 57,595,205 sequences and 20,795 families. PLAID is fully compatible with larger sequence databases such as UniRef or BFD (about 2 billion sequences), however, this study still chose to use Pfam because its sequence domain contains more structural and functional tags, which makes it easier to perform computer simulation evaluation of generated samples. In addition, the researchers also retained data from about 15% for verification.
The UniRef codes for organisms from the Pfam domain are available from the Pfam-A.fasta file provided by the Pfam FTP server. The researchers analyzed all unique organisms in the dataset and found a total of 3,617 different organisms, which they then tested to validate the PLAID approach.
Model Architecture
PLAID is a new paradigm for the multimodal, controllable generation of proteins by diffusion in the latent space of predictive models.The method overview is shown in the figure below. In short, it is divided into 4 steps:
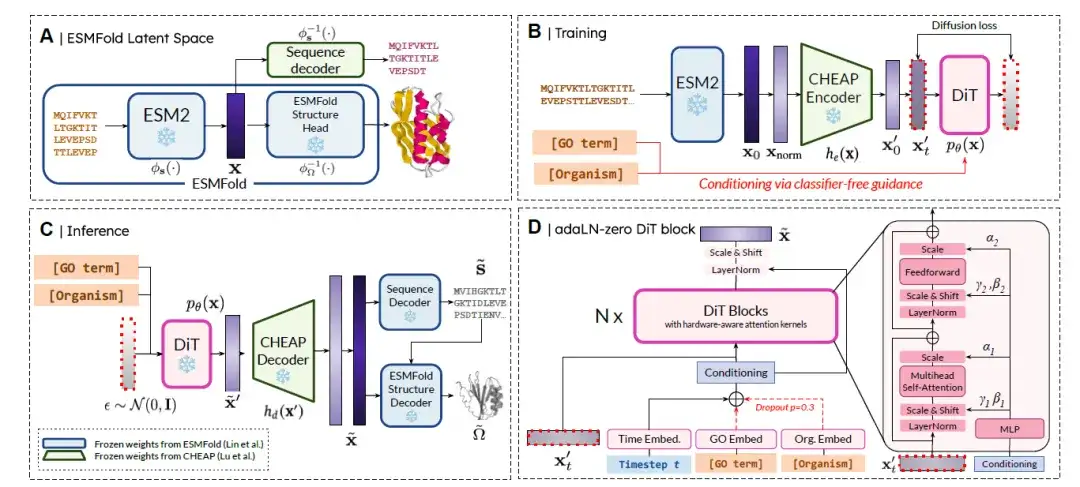
(A) ESMFold Latent Space:The latent space p(x) represents the joint embedding of sequence and structure.
(B) Potential Diffusion Training:The goal is to learn and sample from pθ(x), following the diffusion formula. To improve learning efficiency, the researchers use the CHEAP encoder he(·) to obtain the compressed embedding x′ = he(x), so that the diffusion goal becomes sampling from pθ(he(x)).
(C) Inference:To obtain both sequence and structure at inference time, the researchers used the trained model to sample ˜x′ ∼ pθ(x′), which was then decompressed using the CHEAP decoder to obtain ˜x = hd(˜x′). This embedding is decoded into the corresponding amino acid sequence using a frozen sequence decoder trained in CHEAP. The residue identity sequence and ˜x are used as input to a frozen structure decoder trained in ESMFold to obtain the full-atom structure.
(D) DiT block architecture:The researchers used the Diffusion Transformer (DiT) architecture combined with the adaLN-zero DiT block to fuse conditional information. Functions (i.e., GO terms) and organism class labels were embedded using classifier-free guidance.
Study Results
The researchers conducted a structural quality and diversity analysis of different protein lengths, and the results are shown in the figure below.The native protein and PLAID-generated samples have consistent metrics at different lengths.ProteinGenerator and Protpardelle showed mode collapse at certain lengths, while Multiflow showed a decrease in diversity on longer sequences.
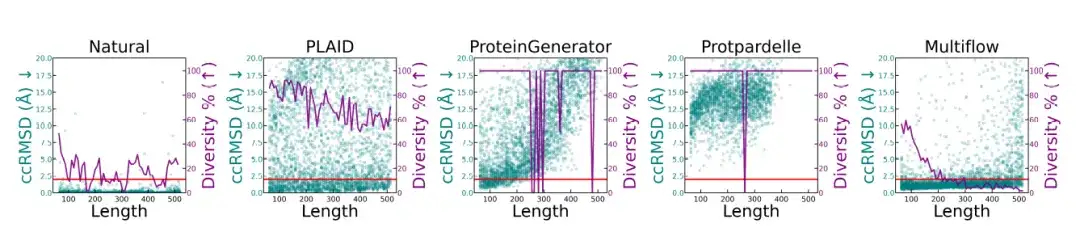
* This figure compares natural proteins and different generation methods, showing the structural quality (ccRMSD, cyan points) and diversity (purple line, measured as the proportion of unique structural clusters in the total sample) of proteins at different lengths (64-512 residues). The red line is at 2Å, indicating the design threshold)
In addition, compared with the baseline method,The secondary structure diversity generated by PLAID more closely resembles the distribution of native proteins.As shown in the figure below: ProteinGenerator, Protpardelle and Multiflow show deviations in their secondary structure distributions, and existing protein structure generation models usually have difficulty generating samples with high β-sheet content.
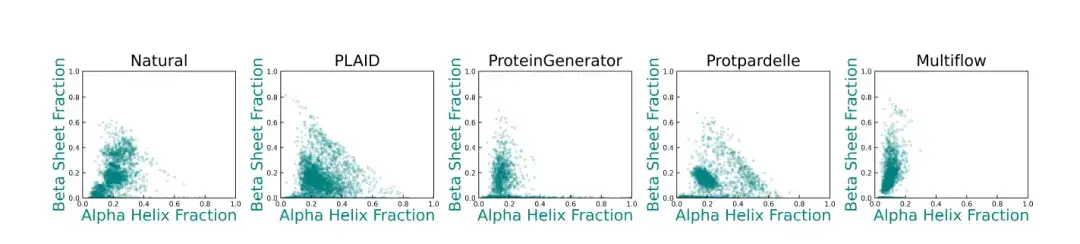
* This figure shows the distribution of α-helical and β-sheet content of natural proteins and protein structures generated by different methods. Each point represents a structure, and its coordinates represent the proportion of α-helical residues (x-axis) and the proportion of β-sheet residues (y-axis)
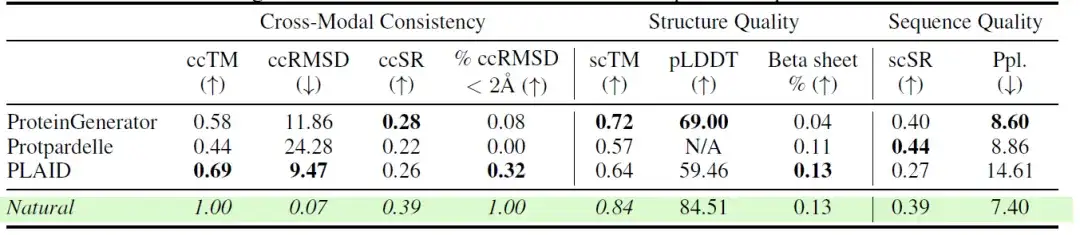
The researchers also compared the performance of different models across multiple consistency and quality metrics in the all-atom protein generation task. The results are shown in the following table:The samples generated by PLAID show high cross-modal consistency between sequence and structure.
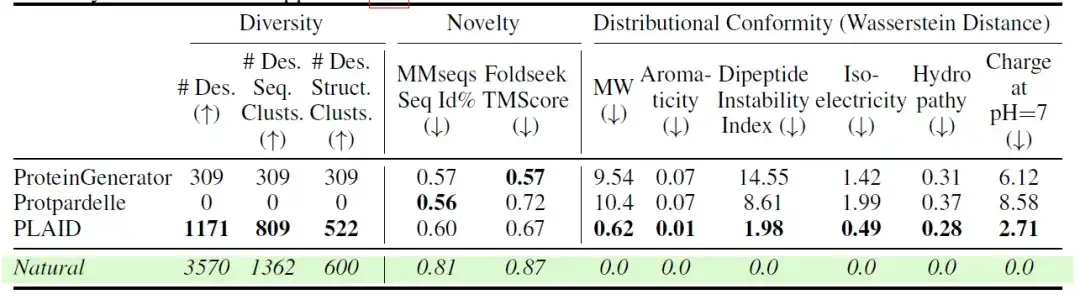
The researchers also further evaluated the diversity, novelty, and naturalness of different models. The results are shown in the following table:Among all-atom models, PLAID generated the most unique and designed samples in both sequence and structural space.
It is worth emphasizing that PLAID can be easily extended to many downstream functions and is not limited to ESMFold, but can be applied to any prediction model.
AI opens up a new avenue for protein research
Diffusion Transformer is increasingly used in the biological field
This paper mentions that during the model building process, the researchers used Diffusion Transformer (DiT) to perform denoising tasks.
The basic principle of DiT is to apply the Transformer architecture to the diffusion model. Diffusion models usually corrupt the original data by gradually adding noise, and then restore the data through model learning. DiT enhances the generative ability of the model by introducing Transformer blocks (such as adaptive layer normalization, cross attention, etc.) into the diffusion model.
In recent years, DiT has made significant progress in the field of image and video generation. The main architecture of cutting-edge generation models such as Sora is DiT.In the field of biomedicine, the application of Diffusion Transformer is becoming more and more extensive. It can help researchers quickly screen potential drug molecules and predict their biological activity. It can also assist in complex tasks such as gene sequence analysis and protein structure prediction, providing a powerful tool for life science research.Taking protein denoising as an example, DiT can capture complex sequence-structure relationships. That is, through the global self-attention mechanism of Transformer, it can effectively model the complex interactive relationship between protein sequence and structure, and then use the inverse process of the diffusion model to predict the denoised latent vector at each time step, gradually restoring the structure and sequence of the protein from the noise.
Specifically in this paper, DiT provides a more flexible option for fine-tuning to handle mixed input modalities, especially when protein structure prediction models begin to integrate nucleic acids and small molecule ligand complexes. In addition, this approach makes better use of the Transformer training infrastructure.
In early experiments, the researchers also found that allocating available memory to a larger DiT model was more effective than using triangular self-attention. The model was trained using the optimization algorithm implemented by xFormers, and in the benchmark test of the inference phase, a speed increase of 55.8% and a reduction in GPU memory usage of 15.6% were obtained.
Machine learning makes customized proteins a “dream come true”
The above-mentioned UC Berkeley research can be said to be another important step in protein customization. We know that proteins are usually composed of 20 different amino acids, which can be regarded as the building blocks of life.Due to its extremely complex structure, decades ago, it was still a "pipe dream" for scientists to predict the three-dimensional structure of proteins and design new proteins for human use. However, the rapid progress of machine learning in recent years has made the dream of designing customized proteins gradually possible.
In addition to the well-known AlphaFold, some research progress is also worthy of attention:
In November 2024, a team from the U.S. Department of Energy's Argonne National Laboratory successfully developed an innovative computing framework called MProt-DPO.This framework combines artificial intelligence technology with the world's top supercomputers, marking a new era in protein design. In actual cases, scientists have designed a new type of enzyme through MProt-DPO, which can efficiently catalyze chemical reactions under specific conditions. Compared with previous design methods, the reaction efficiency of the new enzyme has increased by nearly 30%, which not only speeds up the experimental progress, but also provides more possibilities for industrial applications. In addition, the successful application of MProt-DPO has also opened up new ideas for the design of antiviral proteins. The relevant research results were published in IEEE Computer Society under the title "MProt-DPO: Breaking the ExaFLOPS Barrier for Multimodal Protein Design Workflows with Direct Preference Optimization".
Paper address:
https://www.computer.org/csdl/proceedings-article/sc/2024/529100a074/21HUV88n1F6
Protein pockets are sites on proteins that are suitable for binding to specific molecules. Protein pocket design is one of the important methods in the process of customizing proteins. In December 2024, the University of Science and Technology of China and its collaborators designed the deep generation algorithm PocketGen.The protein pocket sequence and structure can be generated based on the protein framework and the bound small molecules. Experiments show that the PocketGen model exceeds traditional methods in terms of affinity and structural rationality, and has also greatly improved computational efficiency. The relevant research results were published in Nature Machine Intelligence under the title "Efficient generation of protein pockets with PocketGen".
Paper address:
https://www.nature.com/articles/s42256-024-00920-9

In the future, with the further application of artificial intelligence in the field of protein, I believe people will have a deeper understanding of the secrets of protein spatial structure.
References:
1.https://www.biorxiv.org/content/10.1101/2024.12.02.626353v1
2.https://mp.weixin.qq.com/s/_5_L7bvl-vHtls8gBbfSmQ
3.https://mp.weixin.qq.com/s/sfrm2rj_8kH0JA2vu4NmTw
4.http://www.news.cn/globe/20241014/f7137840e56340f081f9eb819d87ba40/c.html
5.http://www.bfse.cas.cn/yjjz/202412/t20241212_5042432.html
6.https://www.sohu.com/a/826241274_12








