Command Palette
Search for a command to run...
Published in Nature Sub-journal! University of California Uses AI to Innovate cryo-electron Microscopy 3D Reconstruction, Achieving a Major Breakthrough in Structural Biology

In the field of scientific research, some technologies often become the focus of the times due to their breakthrough progress. Cryo-electron microscopy (Cryo-EM), which won the 2017 Nobel Prize in Chemistry, is one of such technologies. For example, relying on cryo-EM technology, Shi Yigong's team captured the high-resolution structure of spliceosomes for the first time in 2015. This was hailed as China's greatest contribution to world science in the field of basic life sciences in the past 30 years, and it also attracted widespread attention to cryo-EM.
As an important tool in the field of structural biology, cryo-electron microscopy can quickly cool samples to low temperatures, preventing the crystallization of water molecules in the samples, thereby preserving the near-physiological state of the samples.Once samples are frozen, researchers can use a range of cryo-EM techniques to visualize the samples in 3D at various resolutions, including near-atomic resolution, to gain a deeper and more comprehensive understanding of the samples.
However, although cryo-EM technology has become increasingly mature, the problem of orientation advantage in sample preparation has always been a difficult problem. Generally speaking, the 3D reconstruction process requires protein projections from all directions to cover the entire space. However, proteins adsorbed on the air-liquid interface (AWI) often show orientation advantages, resulting in incomplete projection data sets, which in turn causes varying degrees of distortion in protein density, leading to reconstruction distortion.
recently,The UCLA research team proposed a self-supervised deep learning method called single-particle IsoNet (spIsoNet).This method provides a new way to restore sample isotropy. When spIsoNet is applied to single-particle cryo-EM, it can significantly improve the quality of biological macromolecule reconstruction, enhance alignment accuracy and angular isotropy, and bring new breakthroughs to the field of structural biology.
The research, titled "Overcoming the preferred-orientation problem in cryo-EM with self-supervised deep learning", has been published in the international academic journal Nature Methods.
Research highlights:
* This study developed an end-to-end method based on self-supervised deep learning, spIsoNet, which can be used to improve the image quality of cryo-EM
* spIsoNet can solve the 3D reconstruction problem caused by the preference orientation problem
* spIsoNet improves angular isotropy and particle alignment accuracy during 3D reconstruction

Paper address:
https://doi.org/10.1038/s41592-024-02505-1
spIsoNet dataset address:
https://go.hyper.ai/P7XQu
The open source project "awesome-ai4s" brings together more than 100 AI4S paper interpretations and provides massive data sets and tools:
https://github.com/hyperai/awesome-ai4s
Dataset: Select multiple datasets, each with different characteristics and application scenarios
In this study, the researchers used multiple datasets to test the performance of spIsoNet, each with its own unique characteristics and application scenarios:
* β-Galactosidase Dataset:It contains two subsets with specific orientations, namely 1,513 side-view particles and 950 top-view particles, which are used to verify whether spIsoNet can improve the image quality (map quality) affected by the preferred orientation.
* HA trimer tilted dataset (EMPIAR-10097):It is obtained through a grid tilting strategy, which provides a tilted viewing direction and can be used to evaluate the ability of spIsoNet to handle tilted samples.
* Non-skewed HA trimer dataset (EMPIAR-10096):It was collected under gridless tilt conditions, and by importing 130,000 particles and performing misalignment correction, the final image was obtained at 3.45Å resolution, which can be used to compare the differences in the treatment effects of tilted and non-tilted samples.
* Asymmetric Ribosome Dataset (EMPIAR-10406):It contains the 70S ribosome of the A. baumannii pathogen complexed with amikacin and was used to evaluate the performance of spIsoNet in dealing with complex biomolecular structures.
* HIV VLP Tomography Dataset (EMPIAR-10164):It contains immature HIV-1 dMACANC virus-like particles (VLPs) at a resolution of 3.6 Å. In this study, it provides an in-depth look at the virus particle structure.
spIsoNet: Based on the U-net deep learning model, it consists of two major modules
The neural network used by spIsoNet is based on the U-net network architecture.This is a deep learning model that has been widely recognized in biomedical image restoration and segmentation. As shown in Figure b below, U-net is constructed based on the encoder-decoder architecture by stacking convolutional blocks.
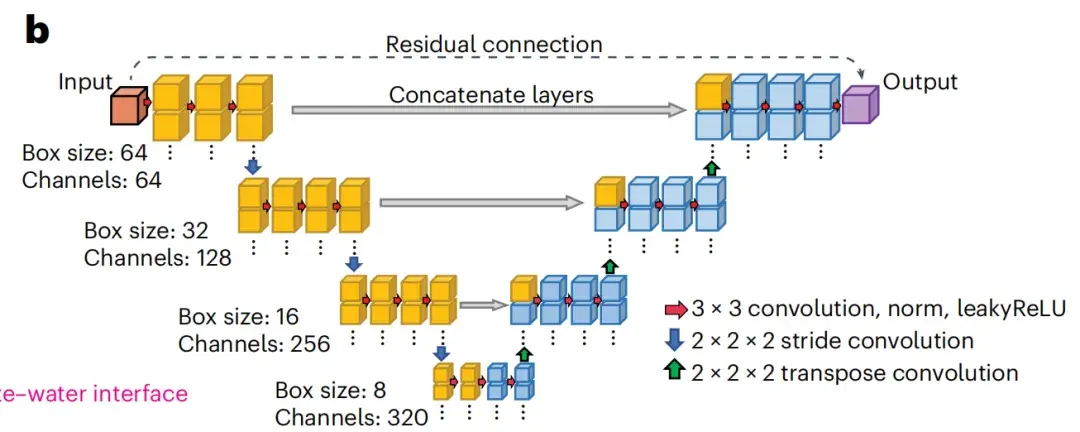
Based on the U-net model, spIsoNet mainly consists of two modules:
The Anisotropy Correction module
The researchers designed an anisotropy correction module to improve the clarity of cryo-electron microscopy images.As shown in Figure c below, the operation of this module takes two halfmaps, a three-dimensional Fourier shell correlation (3DFSC) volume and a solvent mask as input data, and integrates the 3DFSC algorithm to minimize the weighted sum of four different types of loss functions, including consistency loss, equivariance loss, noise-to-noise consistency loss and noise-to-noise equivariance loss, thereby improving the quality of cryo-EM images.
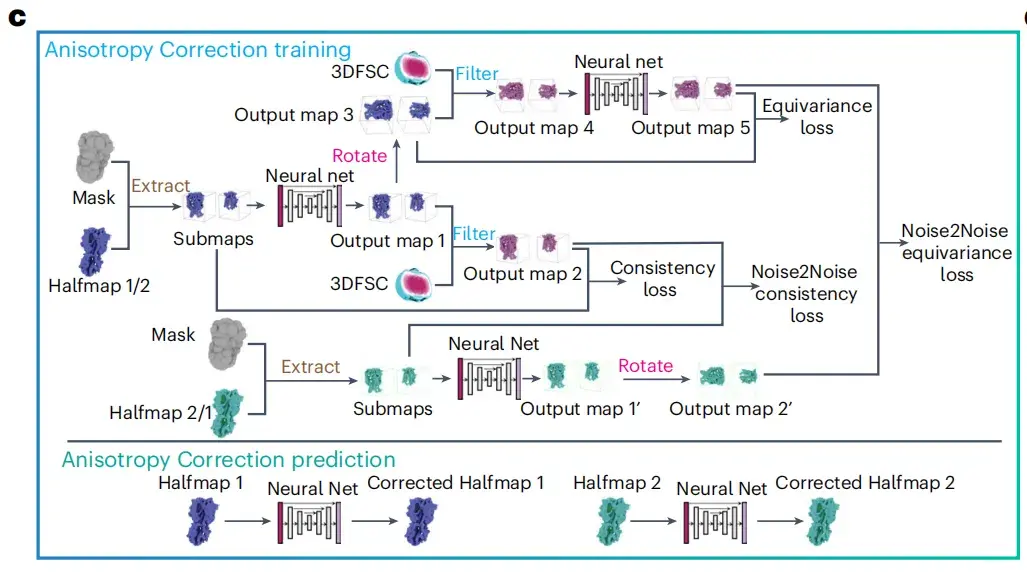
The Anisotropy Correction-powered Misalignment Correction module
As shown in Figure e below, this module integrates a workflow including three main steps: map filtering, anisotropy correction, and RELION auto-refine. Anisotropy correction is the core of the entire process, which aims to improve the quality of cryo-EM images through anisotropy correction.
* Anisotropy correction refers to correcting the differences in the physical and chemical properties of an object in different directions through specific algorithms to achieve an isotropic effect.
* Misalignment correction technology is mainly used to correct image misalignment problems caused by geometric distortion during the imaging process.
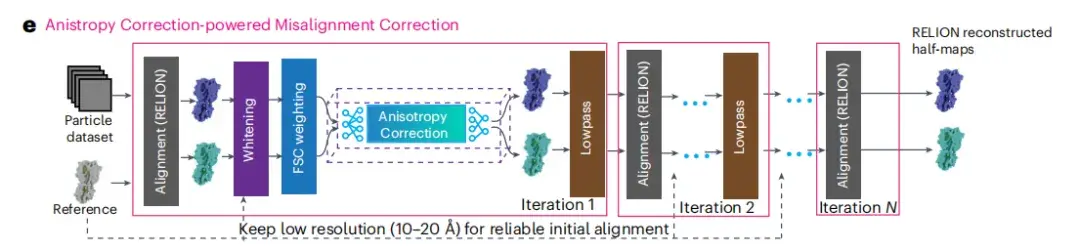
After completing the anisotropy correction, the researchers obtained more accurate particle orientation parameters and two half-images reconstructed by RELION. After each 3D refinement iteration, these half-images are processed by post-processing filters including whitening and FSC weighting to further improve the image quality. The spIsoNet anisotropy correction module then processes these filtered half-images, and the processed corrected half-images are low-pass filtered to meet the standards that match their resolution. These two filtered and corrected half-images will be used as a reference for subsequent orientation estimation.
Research results: spIsoNet significantly improves cryo-EM image quality
Anisotropy correction is effective
The researchers found that the anisotropy correction module of spIsoNet can effectively restore the missing information in the simulated data.The study first tested spIsoNet on the RELION tutorial dataset containing β-galactosidase.
As shown in Figure jm below, by selecting side view particles and top view particles from the 2D class average, the researchers sorted out two subsets of particles with preferred orientations and performed standard RELION 3D reconstruction. The test results show that the anisotropy correction module alone can effectively reduce 3D reconstruction artifacts caused by top view-dominated or side view-dominated orientations.
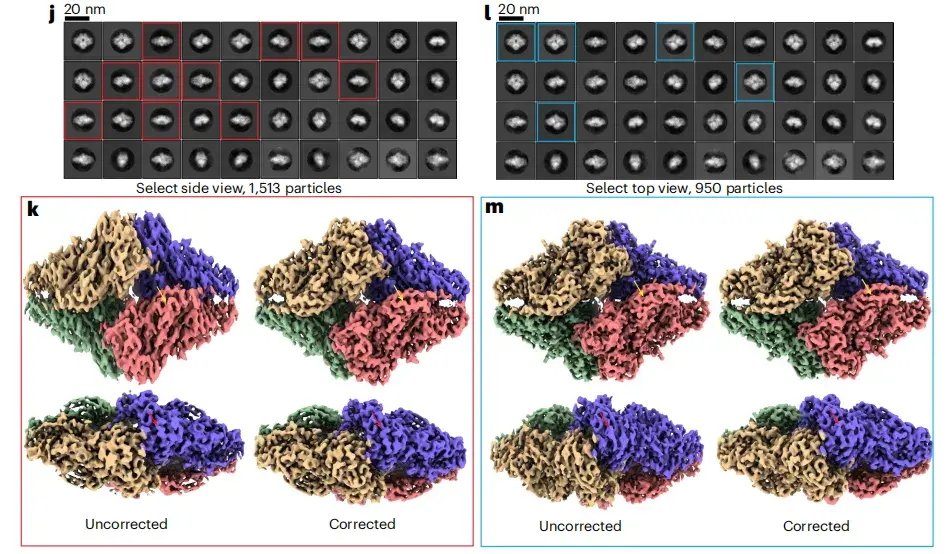
* Where jl refers to the 2D classification map reconstructed from different angles, and km refers to the 3D classification map reconstructed from different angles.
Anisotropy correction and misalignment correction techniques significantly improve cryo-EM image quality
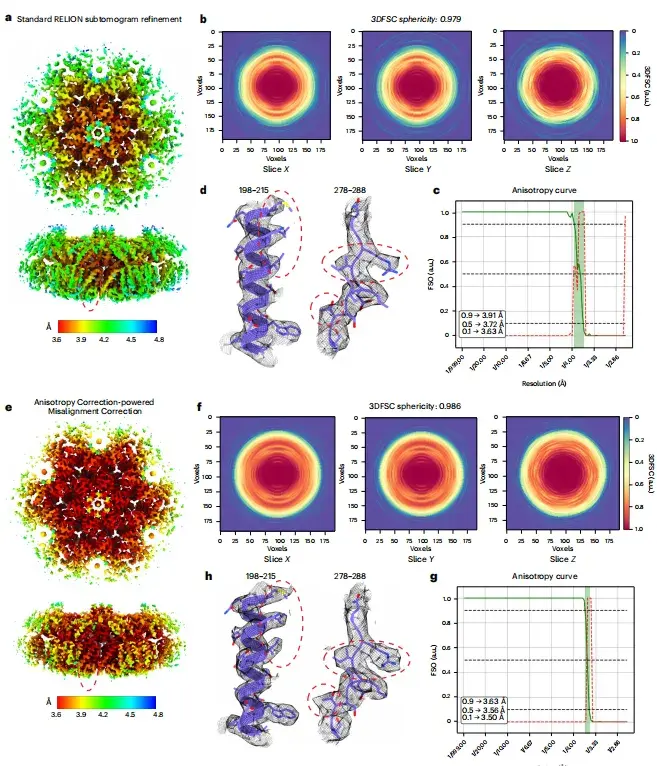
Previous studies have shown that the quality of cryo-EM images of HA trimer tilted datasets is not ideal. To test the effect of spIsoNet,This study first performed anisotropic correction on the halfmap.The results show that the quality of the corrected image is significantly improved, the local resolution is improved, and the noise is reduced. As shown in Figures ab below, in the corrected image, the side chain density that was previously difficult to identify in the original image becomes clearly visible.
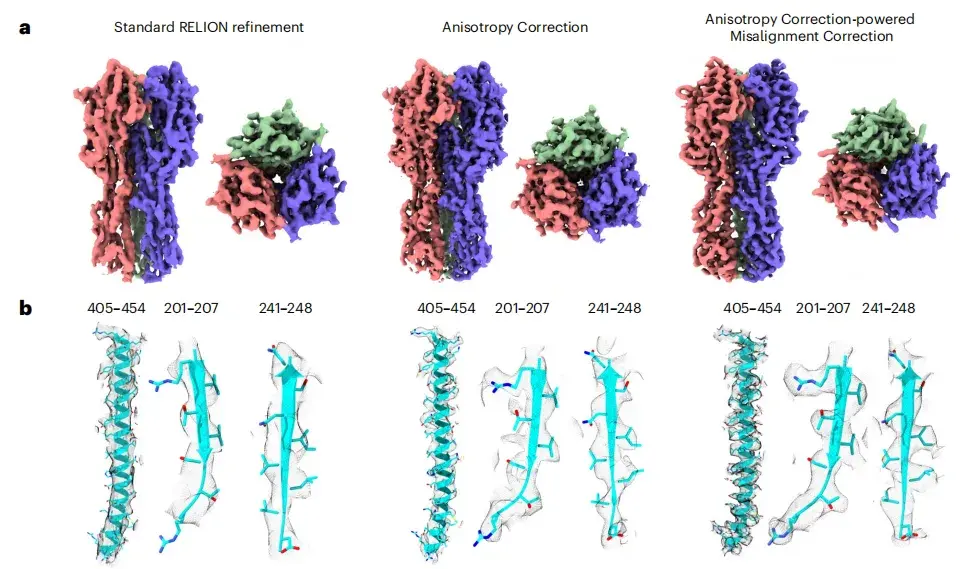
* From left to right: standard RELION refinement, anisotropy correction, and misalignment correction driven by anisotropy correction.
Furthermore, as shown in Figure cf below, the image after deflection correction improves the Fourier shell correlation from image to model, and the three-dimensional Fourier shell correlation (3DFSC) is close to spherical (0.991). Compared with the original image, the image after correction also shows a larger isotropic Fourier shell occupied area (FSO).
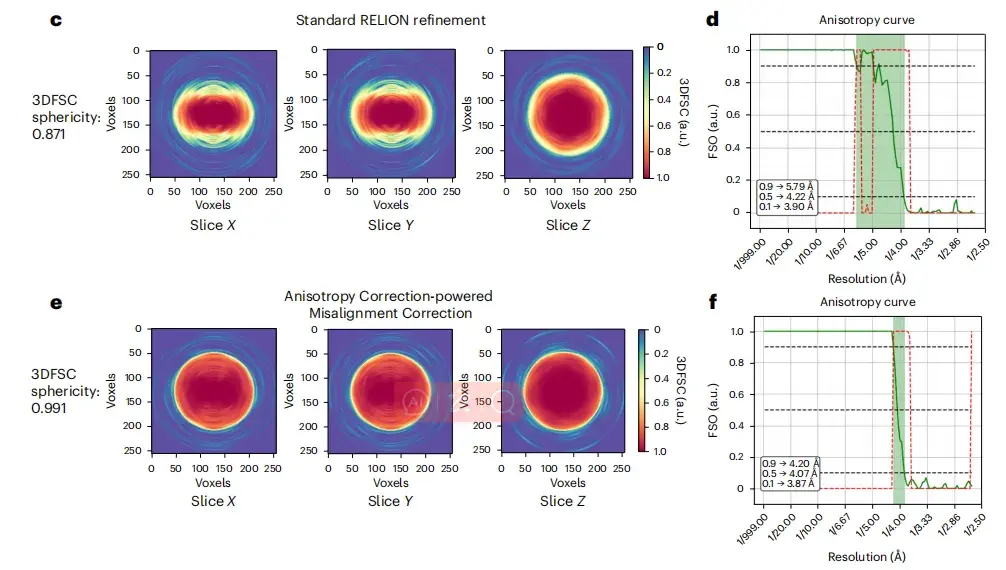
* Where, ce are the 3DFSC slices used for RELION refinement and spIsoNet misalignment correction, respectively, and df are the FSO and P values of Bingham test calculated from the RELION refinement and spIsoNet misalignment correction results, respectively.
Misalignment correction successfully identified and corrected many incorrectly assigned directions
For a protein dataset with serious bias orientation problems, the non-tilted HA trimer dataset (EMPIAR-10096)This study used the anisotropy-correction-driven misalignment correction module of spIsoNet to process particle datasets.The reconstructed HA trimer image in the tilted dataset was used as the reference model.
After misalignment correction, as shown in Figures b-f below, the researchers obtained an image with the correct shape and a significant improvement in isotropy. As shown in Figure h below, the image resolution determined by the half-map FSC (3.5Å) and the model-to-map FSC (3.6Å) was consistent.
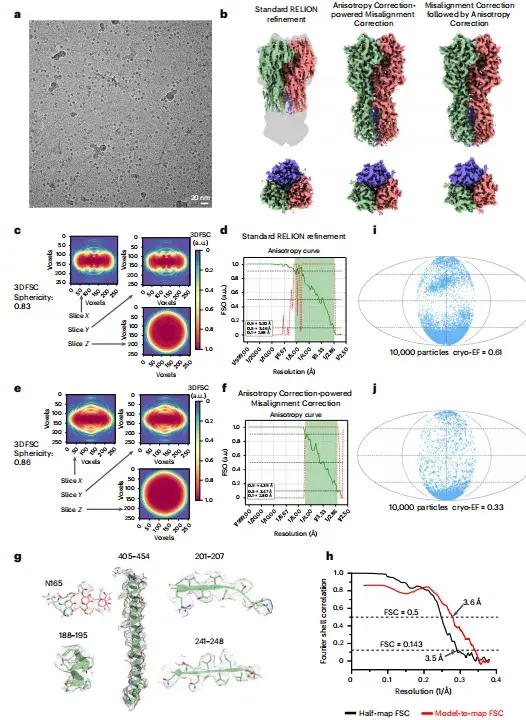
*a-representative cryo-EM micrographs, b-cryo-EM images of HA trimer reconstructed by different methods, c-slices of 3DFSC used for standard RELION refinement, d-FSO and P-values of Bingham test calculated based on standard RELION refinement results, e-slices of 3DFSC corrected by spIsoNet misalignment, f-FSO and P-values of Bingham test calculated based on spIsoNet misalignment correction results, g-representative density of selected amino acid residues and glycans from cryo-EM images, h-corrected FSC curve of HA trimer, i,j-distribution results in different directions and corresponding cryoEF scores
spIsoNet excels in improving alignment of asymmetric particles and particles containing nucleic acid molecules
As shown in Figures ad below, after anisotropy correction, the image quality is significantly improved, showing a more continuous density distribution, higher local resolution, and less noise interference.The study found thatWhen local subtomogram averaging of 70S or 80S ribosomes is used as a reference and the initial resolution of 15Å is maintained for alignment, high-quality images without model bias can be consistently obtained and the effects of anisotropy can be effectively mitigated.
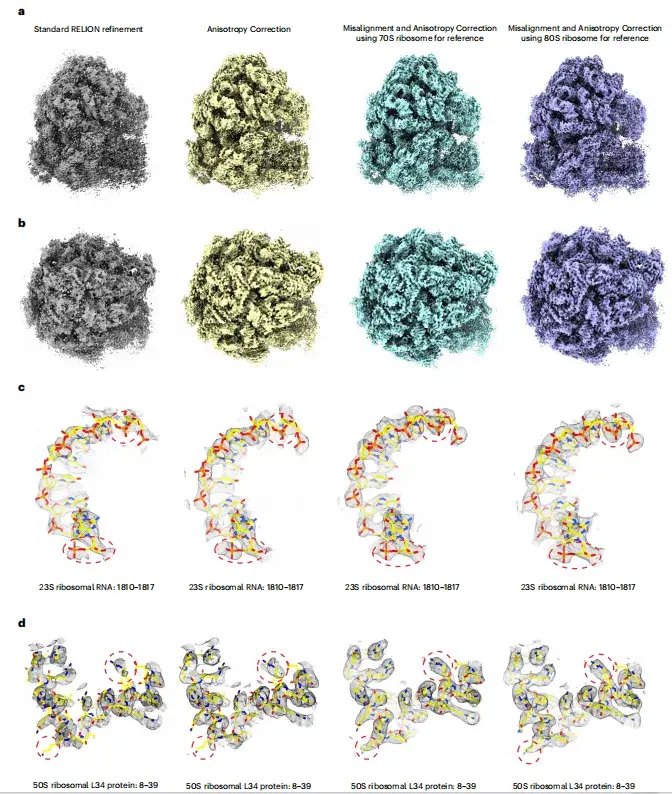
* a, b- ribosome images reconstructed by different reconstruction methods, c, d- representative density regions with fitted atomic models (yellow)
spIsoNet has potential for application in in situ structural biology
To evaluate the application of spIsoNet in subtomogram averaging, this study used the HIV-1 VLP tomography dataset (EMPIAR-10164) as an example.
As shown in Figure a below, in the standard workflow of RELION4, the study used a subset of 5 different tilt angles to obtain a 3.7Å resolution structure. Then, by implementing misalignment correction, the researchers were able to obtain an isotropic 3.6Å resolution structure as shown in Figure e below.
As shown in Figures bh below, the structural analysis further reveals clearer side chain density and exhibits higher 3DFSC sphericity in the FSO curve, which helps to improve the accuracy of particle orientation estimation.
* a- HIV-1 local resolution map reconstructed according to standard RELION, b- slice of 3DFSC used for standard RELION refinement, c- FSO and P-value of Bingham test calculated according to standard RELION refinement results, d- representative density of amino acid residues and glycans selected from cryo-EM images, e- HIV-1 local resolution map reconstructed according to spIsoNet anisotropy correction technology, f- slice of 3DFSC corrected by spIsoNet anisotropy, g- FSO and P-value of Bingham test calculated according to spIsoNet anisotropy correction results, h- representative density of amino acid residues and glycans selected from cryo-EM images
AI+cryo-electron microscopy, a technological paradigm of “strong combination”
In the past two years, there has been a controversial topic in the scientific community: "Does AlphaFold end structural biology?" The answer is of course no.
on the one hand,The training data for structure prediction models such as AlphaFold comes from traditional structure analysis methods such as X-rays and cryo-electron microscopy.on the other hand,Cryo-EM technology excels in analyzing protein dynamics, which is currently not possible with AlphaFold. So, can AI technology represented by AlphaFold assist traditional methods represented by cryo-EM? This can be said to be inevitable.
For example, as early as 2022,Professor Mao Youdong's team at Peking University used AI + cryo-electron microscopy toThe team successfully captured the instantaneous conversion of the induced proteasome from the intermediate state of substrate degradation to the intermediate state of substrate inhibition. This is the first time in the world that artificial intelligence four-dimensional reconstruction technology has been applied to improve the analytical accuracy of time-resolved cryo-EM. The team has achieved atomic-level functional dynamics observation of target protein complexes related to major diseases. The relevant results were published in Nature under the title "USP14-regulated allostery of the human proteasome by time-resolved cryo-EM".
Paper link:
https://doi.org/10.1038/s41586-022-04671-8
Not long ago,Researchers from ByteDance Research team proposed a new method called CryoSTAR.This is the first method to apply protein atomic structure modal priors to cryo-EM experimental data. It uses atomic model information as structural regularization to clarify the conformational heterogeneity of biological macromolecules. It can output coarse-grained models and density maps to show the conformational changes of molecules at different levels, significantly improving the application potential of cryo-EM in dynamic conformational analysis. The related results were published in Nature Methods under the title "CryoSTAR: Leveraging Structural Prior and Constraints for Cryo-EM Heterogeneous Reconstruction".
Paper link:
https://www.nature.com/articles/s41592-024-02486-1
There is no doubt that the combination of AI and cryo-electron microscopy is opening a new chapter in structural biology and also demonstrates the great potential of AI technology in assisting traditional structural biology methods.








