Command Palette
Search for a command to run...
Share NeurIPS 2024 Submission Experience! Zhejiang University Team Uses DePLM Model to Assist Protein Optimization, and the First Author of the Paper Demonstrates the Demo Online

Harry Shum, a foreign member of the U.S. National Academy of Engineering, once emphasized: "If there is something we must do today, it is AI for Science. It is hard to imagine that there is anything more important today, and the awarding of this year's Nobel Prize is the best proof of this."
In the past, scientists relied on manual data organization and hypotheses based on subject theories. Now, with the assistance of AI, research is conducted directly based on massive data. AI for Science has not only improved scientific research efficiency, but also changed the entire scientific research paradigm, which is particularly evident in the field of protein research.
In the fifth episode of Meet AI4S, HyperAI was fortunate to invite Wang Zeyuan, a doctoral student from the Knowledge Engine Laboratory of Zhejiang University,He gave a detailed introduction to a paper of the team selected for NeurIPS 2024, titled "Using the diffusion denoising process to help large models optimize proteins" "DePLM: Denoising Protein Language Models for Property Optimization".
As the top conference in the field of AI, NeurIPS is known as one of the most difficult, highest-level and most influential AI academic conferences. This year, the conference received a total of 15,671 valid paper submissions, an increase of 27% over last year, but the final acceptance rate was only 25.8%. The selected papers are of great learning value.In this sharing session, Dr. Wang Zeyuan introduced in detail the design concept, experimental conclusions, demo operation mode and future prospects of the denoising protein language model DePLM. In addition, he also shared his experience in submitting papers to top conferences, hoping that it will be helpful to everyone.
Specifically, Dr. Wang said that when we submit papers, we can start from topic selection, innovative points, paper writing, and coping with interdisciplinary reviews.
First, in terms of topic selection,You can read a wide range of papers from top conferences to understand the research directions that are of interest to the community. For example, when preparing the DePLM paper, Dr. Wang found that protein engineering, especially protein prediction tasks, was a hot topic at last year's ICLR and NeurIPS conferences.
Secondly, in terms of innovation,He believes that it is important to cultivate the ability to discover problems. In the field of AI for Science, we should first have an in-depth understanding of the knowledge in the field of Science, and compare it with the content in the field of AI to find out the blank areas that have not yet been explored by AI.
In terms of essay writing,He said that writing must be logically clear and detailed to ensure that the article is easy to understand. It is also necessary to communicate more with tutors and classmates to avoid falling into one's own fixed thinking patterns.
Finally, considering that AI for Science papers may be reviewed by reviewers from two different backgrounds, one focusing more on AI technology and the other focusing on Science applications,Therefore, it is necessary to clarify the core positioning of the paper when writing.That is, whether this paper is aimed at the AI community or the Science community, and a logical framework is constructed accordingly to ensure that the content is closely related to the topic.
In his opinion, the current trend of large-scale model research has changed. We have shifted from simple copycat approach to a deep understanding of large-scale models.In the past, we let large models adapt to various downstream tasks, but now we are more concerned about how to make downstream tasks better cooperate with the pre-training stage of large models. The higher the fit between the two, the better the performance of the model.
For example, for predicting adaptive landscapes, traditional simple fine-tuning methods perform poorly in generalization, so we need to have a deeper understanding of large models and unsupervised learning paradigms, identify their deficiencies and improve them. In addition, we must also pay attention to the defects of large models themselves, such as exploring ways to eliminate model bias to optimize model performance.
Model open source and testable
Today I would like to share a paper we published at NeurIPS 2024, which explores how to use diffusion denoising models to help large language models optimize proteins.In this paper, we propose a novel Denoised Protein Language Model (DePLM)The core of this approach is to view the evolutionary information captured by the protein language model as a mixture of relevant and irrelevant information to the target feature, where irrelevant information is considered as "noise" and eliminated. We found that the proposed ranking-based denoising process can significantly improve the optimization performance of proteins while maintaining strong generalization capabilities.
Currently, DePLM has been open sourced. Due to the complex configuration environment of the model,We have launched "DePLM: Optimizing Proteins with Denoised Language Models (Small Samples)" in the tutorial section of the HyperAI official website.In order to help you better understand and reproduce our work, I will explain how the model works from several aspects, including how the DePLM model works, what its related configuration files are, how to fine-tune the diffusion steps of the model, and how to run the DePLM model with your own data set.
DePLM open source address:
https://github.com/HICAI-ZJU/DePLM
DePLM tutorial address:
Background: Maximizing the use of evolutionary information and minimizing the introduction of data bias signals
The research object of this paper is protein, which is a biological macromolecule composed of 20 amino acids in series. It undertakes functions such as catalysis, metabolism and DNA replication in the body, and is also the main executor of life activities. Biologists usually divide its structure into four levels. The first level describes how the protein is composed; the second level describes the local structure of the protein, such as the common α helix and β fold; the third level describes the overall three-dimensional structure of the protein; and the fourth level considers the interaction between proteins.
At present, most AI+protein research can be traced back to natural language processing research because there are similarities between the two. For example, we can compare the quaternary structure of protein with letters, words, sentences, and paragraphs in natural language. When a letter error occurs in a sentence, the sentence loses its meaning. Similarly, mutations in the amino acids of a protein may also cause the protein to be unable to form a stable structure and thus lose its function.
As shown in the figure below, in the paper "Controllable protein design with language models", researchers corresponded natural language with proteins. This approach has been widely recognized by researchers. Since 2020, protein research has shown explosive growth.
Original paper:
https://www.nature.com/articles/s42256-022-00499-z
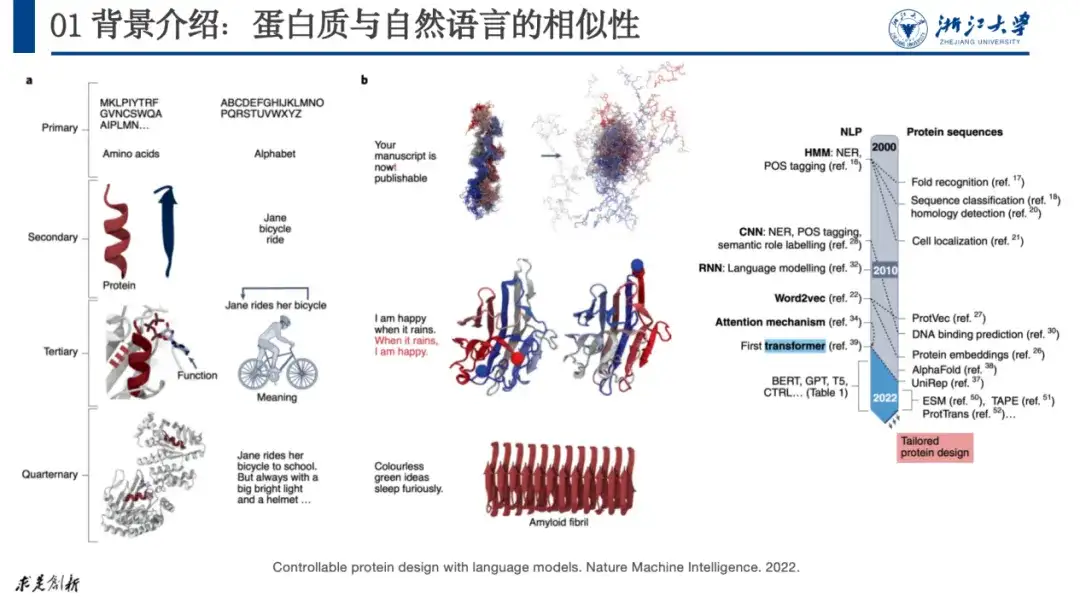
The issue we are discussing this time is AI+protein optimization, that is, if we have a protein that does not function as expected, how to adjust its amino acid sequence to meet the expected function.
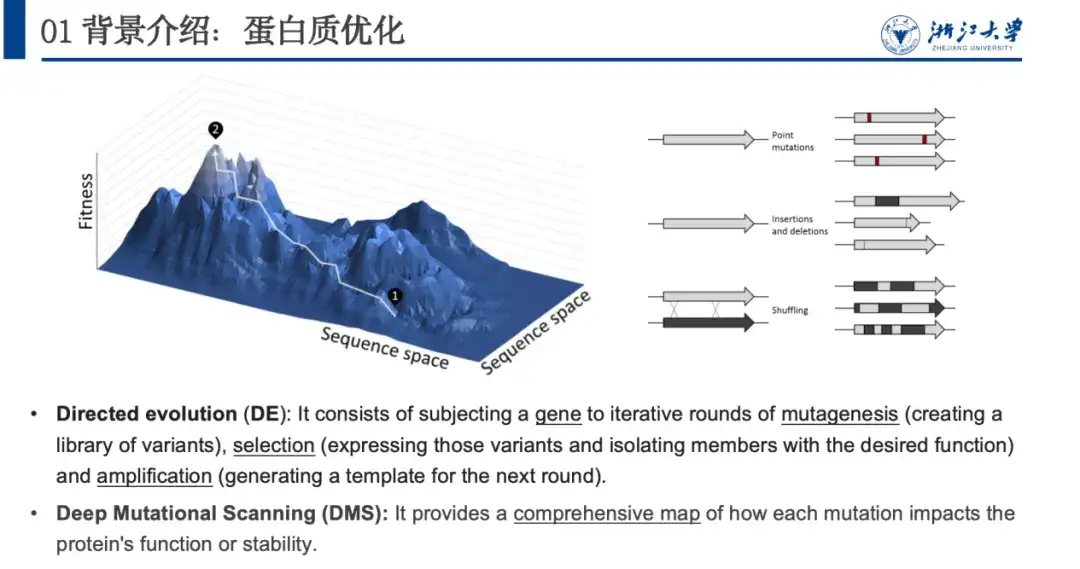
In nature, proteins continuously optimize themselves through random changes (including insertion, deletion or mutation of points). By imitating this process, biologists proposed directed evolution and deep mutation scanning to optimize proteins. The problem with these two methods is that they consume too much experimental resources. Therefore,We use computational methods to model the relationship between proteins and their property fitness, i.e., predict the fitness landscape, which is crucial for protein optimization.
In order to model this problem, we usually use data sets, evaluation metrics, and calculation methods.As shown in the figure below, a protein optimization dataset usually contains a wild-type sequence xwt, multiple mutation pairs μi, and predicted fitness values yi after mutation. The evaluation model mainly relies on the Spearman's correlation coefficient. This indicator does not focus on the specific predicted value, but on the ranking of fitness value changes caused by mutation. The closer the ranking value of the actual mutation R(Y) is to the predicted fitness score, the better the model training is.
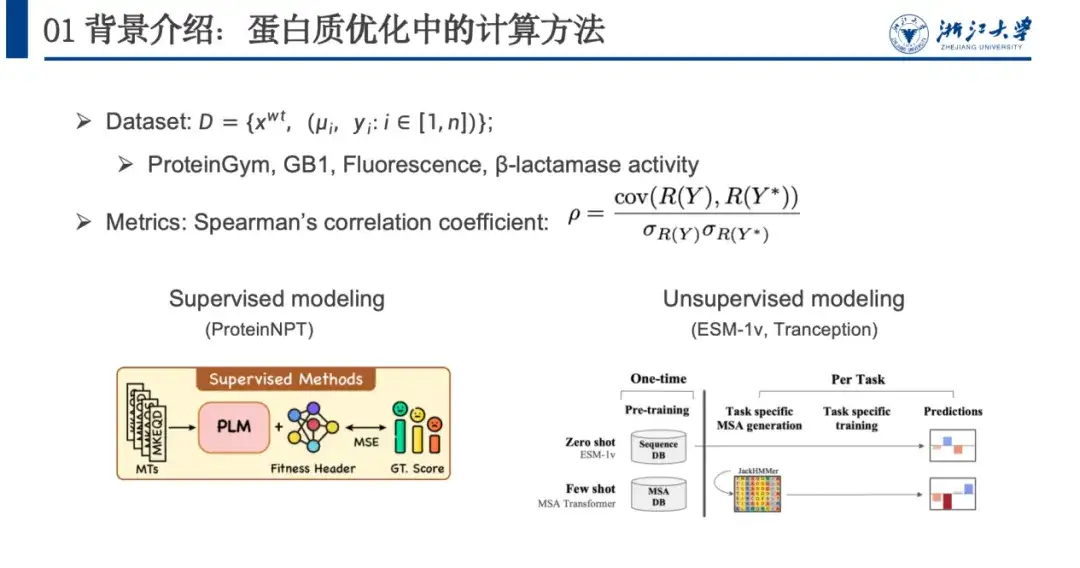
Computational methods can be roughly divided into supervised modeling and unsupervised modeling. Supervised learning relies on labeled data and trains the model by optimizing the loss function to improve the prediction ability of fitness. Unsupervised learning does not require labeled data, but performs self-supervised learning on a large-scale protein dataset that is unrelated to fitness. The model only needs to be trained once and can be generalized to various protein prediction tasks.
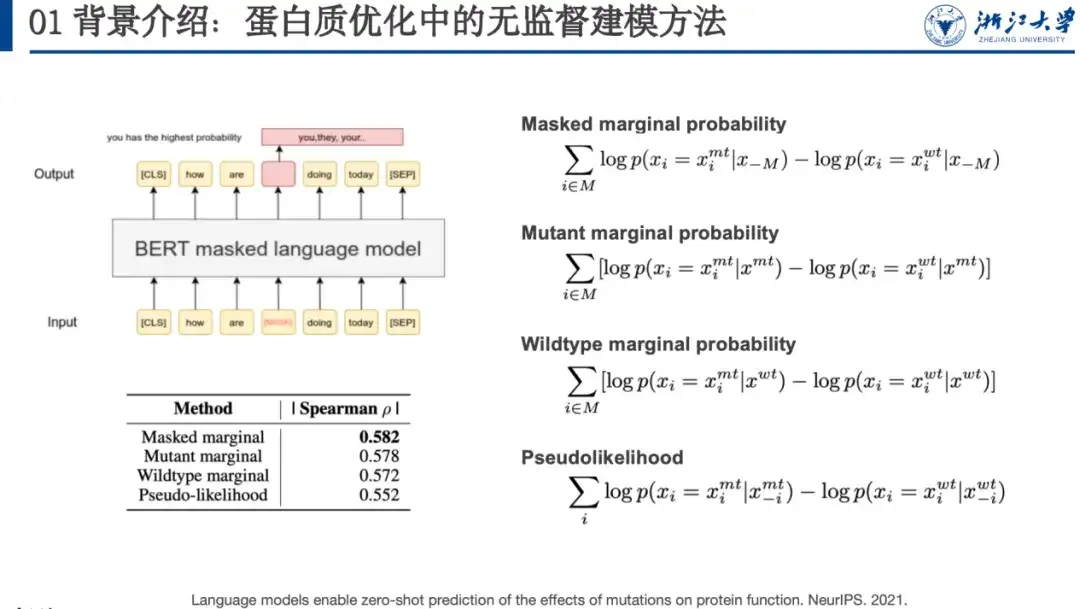
For example, masked language modeling is an unsupervised learning method. When training the model, we need to provide the model with a contaminated sequence. We can mask a word (such as the word in the red box in the figure below) or randomly change it to another word, and let the language model restore it, that is, restore the original sequence. In a paper at NeurIPS 2021, researchers found that the probability of protein mutation predicted by such models is correlated with the fitness landscape. To this end, they designed 4 mutation scoring formulas, as shown on the right side of the figure below.
Original paper:
https://proceedings.neurips.cc/paper/2021/file/f51338d736f95dd42427296047067694-Paper.pdf
In summary, supervised methods perform well but have limited generalization capabilities, while unsupervised methods perform slightly worse but have strong generalization capabilities.In order to combine the advantages of both, as shown in the figure below, we borrowed the Pre-train + Fine-tune strategy in the NLP field. After some attempts, we found that although this method performed well, it had poor generalization ability, similar to supervised learning. So we analyzed why unsupervised methods have excellent generalization ability and assumed that this generalization ability comes from evolutionary information (EI). This is because organisms can optimize proteins through natural evolution, and this evolutionary mutation will also be retained. Therefore, we consider that the correlation between mutation probability and fitness landscape is positively correlated.

However, when we tried to fine-tune the model, we actually used embedding information and did not fully utilize evolutionary information. In addition, there was a bias in irrelevant information in the wet experiment data. We believe that evolutionary information contains comprehensive information in various directions, such as stability, activity, expression, binding, etc. When we optimize the stability of proteins, the evolution of activity, expression, and binding is irrelevant information. If the probability value of this uninteresting information can be removed, the performance of the model can be improved. Because the whole process is carried out in the likelihood space, it will not affect the generalization ability of the model.Therefore, we need to maximize the use of evolutionary information during fine-tuning while minimizing the bias signal introduced into the dataset.
DePLM algorithm framework: denoising model based on sorting space
Based on this, we proposed the DePLM model, the core idea of which is to regard the evolutionary information captured by the protein language model as a fusion of interesting and uninteresting signals, the latter of which is regarded as "noise" in the target attribute optimization task and needs to be eliminated. DePLM denoises the evolutionary information by performing a diffusion process in the sorting space of attribute values, thereby enhancing the generalization ability of the model and predicting mutation effects.
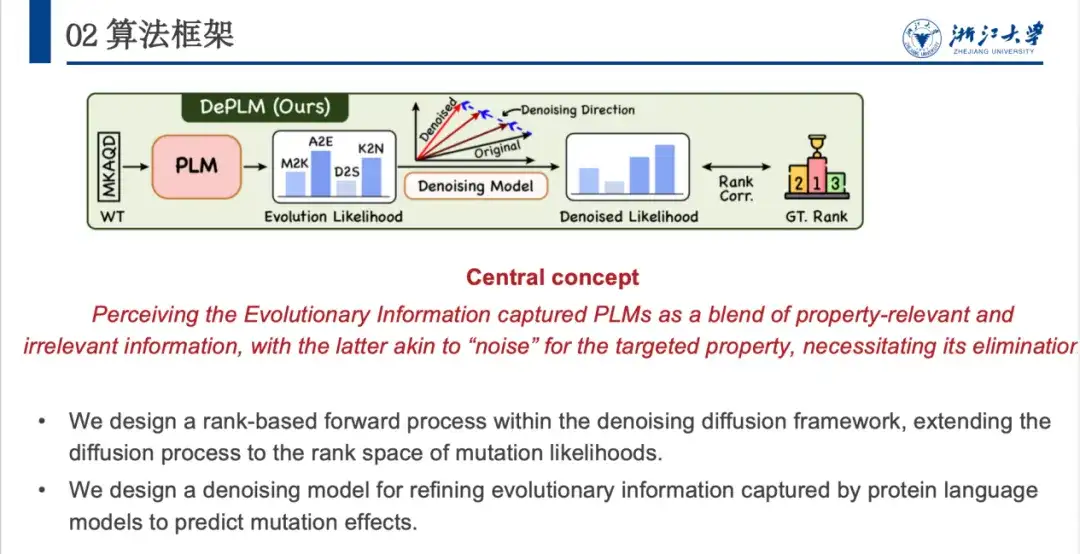
Given an amino acid sequence of a protein, the model predicts the probability of each position mutating into various amino acids, and the evolutionary likelihood then generates the likelihood of the property of interest through the Denosing Module. Specifically,DePLM mainly consists of two parts: the forward diffusion process and the learned backward denoising process.In the forward process, a small amount of noise is gradually added to the true situation, and in the reverse denoising process, learning is done to gradually remove the accumulated noise and restore the true situation.
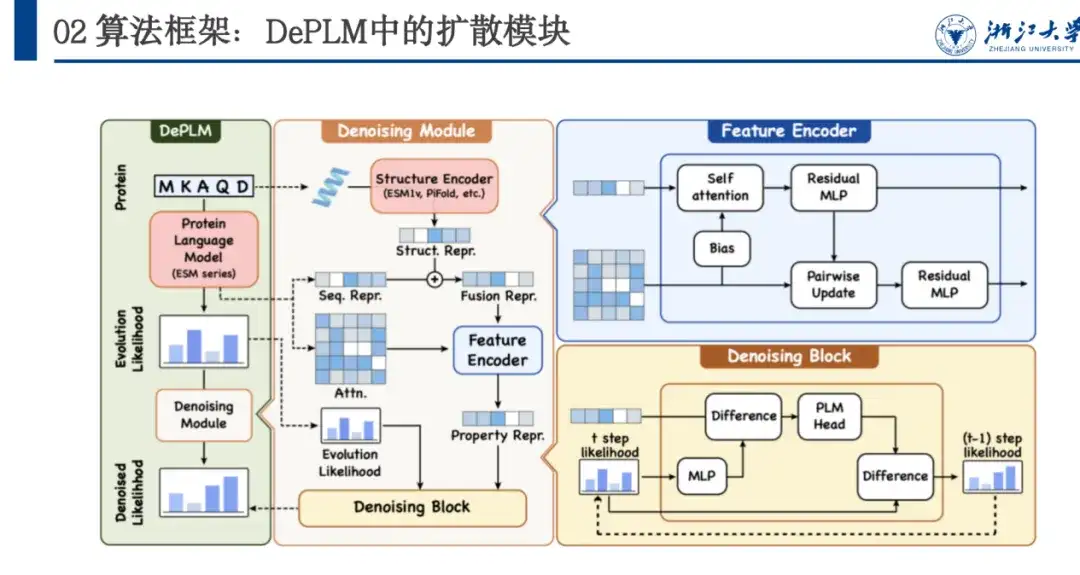
As shown in the figure below, DePLM is based on the ESM series and uses the Transformer architecture. Its Denosing Module is trained based on the diffusion process, and the network architecture includes Feature Encoder and Denosing Block. Feature Encoder extracts sequence features from the Protein Language Model and extracts structural features through the ESM 1v model. These two features are used as anchors, and multiple rounds of Denoising Block iterations are used to gradually denoise and obtain the Denosed likelihood.
In the past, denoising methods were mostly used in the field of image generation, especially in diffusion models. As shown in the figure below, the original image x0 is converted into a noise space (xT) close to a Gaussian distribution through a defined denoising process, and then the model learns the reverse denoising process.
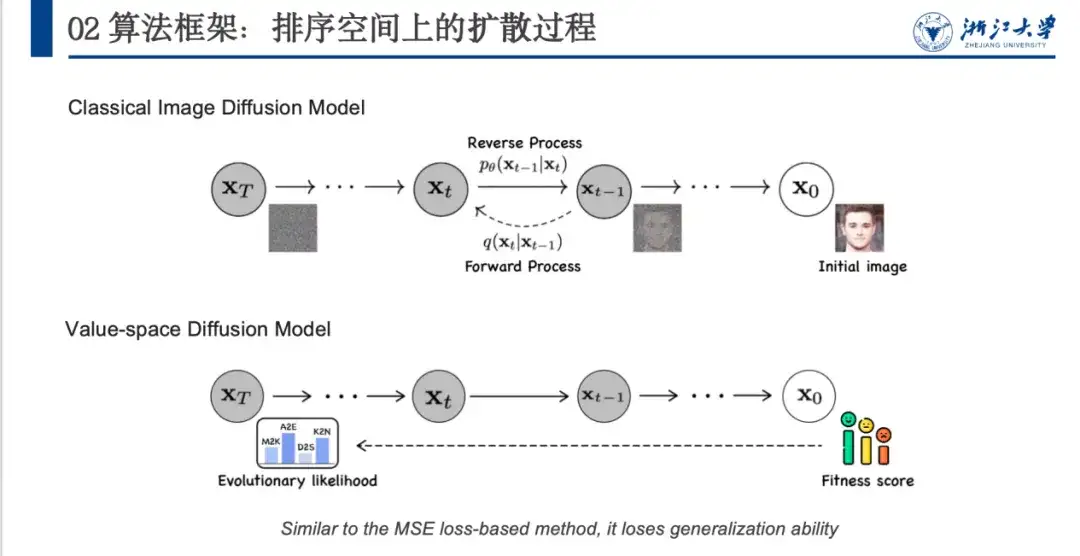
However, there are some problems in directly applying the image denoising model to the protein field. As shown in the figure above, the image denoising model can add random noise to form an inseparable noise space (from x0 to xT). However, proteins have fitness scores and evolutionary likelihoods, and the initial and final states are fixed. Therefore, the noise addition process needs to be carefully designed. Secondly, the model will align to the fitness score, resulting in good performance but poor generalization ability.
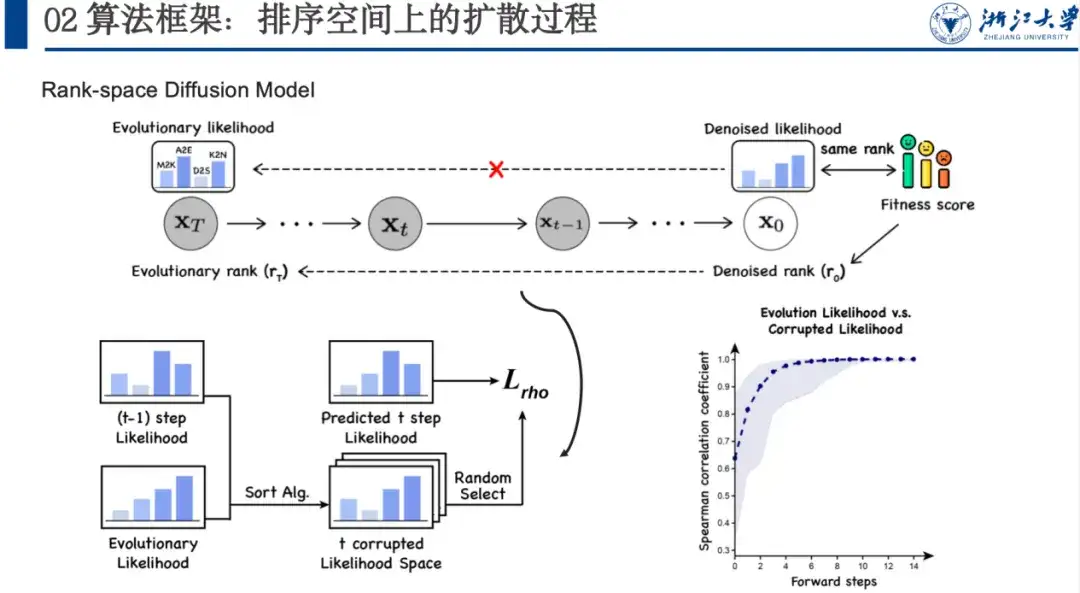
We therefore propose a denoising model based on the ranking space, focusing on maximizing the ranking relevance.This is because we want to denoise the evolutionary likelihood to the property space of interest. Although we do not know the specific situation of this space, we know that its sorting is consistent with the fitness sorting.
We add noise to this space, allowing the model to learn a large number of data sets and gradually learn what the Denosied likelihood should look like, rather than directly aligning the fitness score. In this forward noise-adding process, we use a sorting algorithm to make each step of the sorting closer to the final state and contain randomness, and the model will also learn the reverse step-by-step sorting idea. Specifically, as shown in the figure below, if we have xt-1, we can feed the sorting algorithm with xt-1 and xT to let it sort multiple times. When the sorting space of the tth step is obtained, the sorting variable of the tth step can be randomly sampled from it, allowing the model to predict the likelihood from the t+1 step to the tth step, and calculate the Spearman loss. Since we do not need to add many steps like image denoising, the sorting process can usually be completed in 5-6 steps, which also improves efficiency.
Experimental conclusion: DePLM has superior performance and strong generalization ability
To evaluate the performance of DePLM in protein engineering tasks, we compared it with protein sequence encoders trained from scratch, self-supervised models, etc. on ProteinGym, β-lactamase, GB1, and Fluorescence datasets. The results are shown in the figure below. DePLM outperforms the baseline model. By comparison,We find that high-quality evolutionary information can significantly improve the fine-tuning results, which demonstrates the effectiveness of our proposed denoising training procedure and confirms the advantage of integrating evolutionary information with experimental data in protein engineering tasks.
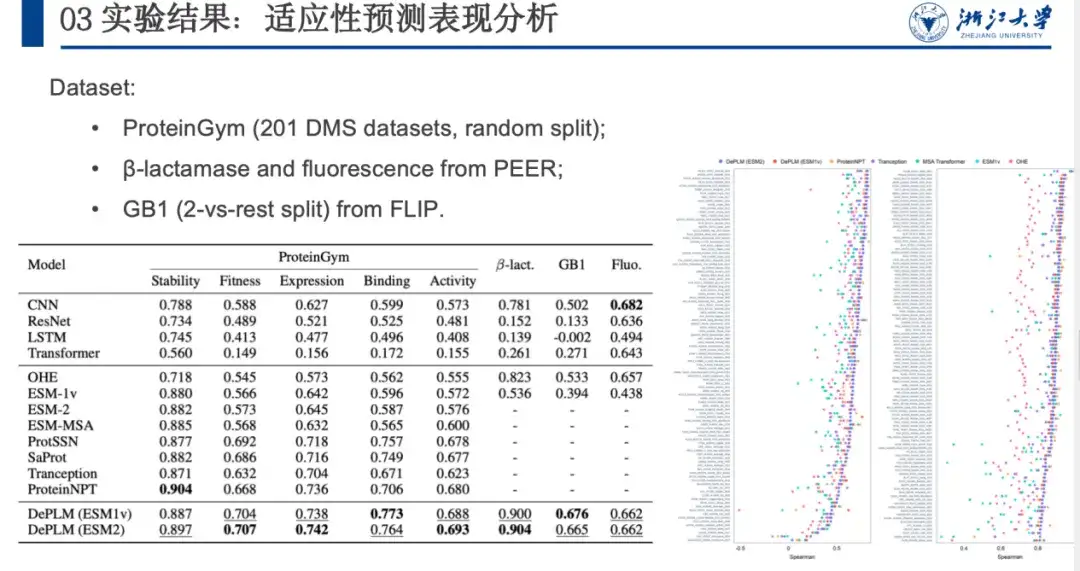
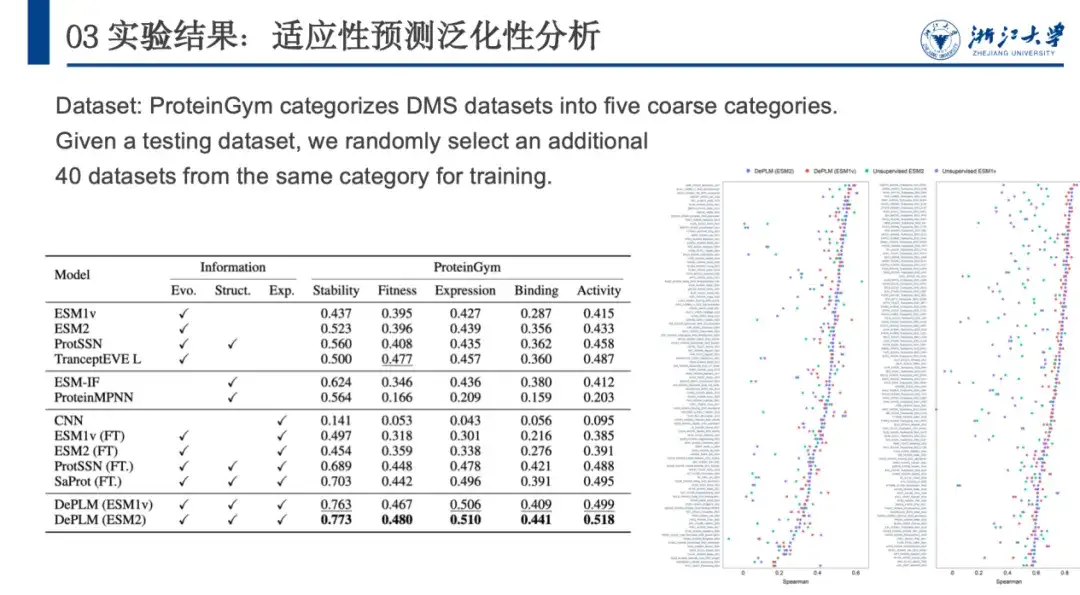
Next, to further evaluate the generalization ability of DePLM, ProteinGym divides the DMS dataset into five categories according to the protein properties they measure, namely stability, fitness, expression, binding, and activity. We compare it with other self-supervised models, structure-based models, and supervised baseline models. The results are shown in the figure below. DePLM outperforms all baseline models.This shows that models that only rely on unfiltered evolutionary information are insufficient, as they often dilute the target attributes due to optimizing multiple objectives simultaneously. By eliminating the influence of irrelevant factors, DePLM significantly improves performance.
To further analyze the generalization performance and determine the importance of filtering out attribute-irrelevant information, we performed cross-validation of training and testing between attributes. As shown in the figure below, in most cases, when the model is trained on attribute A and tested on attribute B, its performance is lower than when it is trained and tested on the same attribute (i.e., A).This shows that the optimization directions of different properties are not consistent and there is mutual interference, which confirms our initial hypothesis.
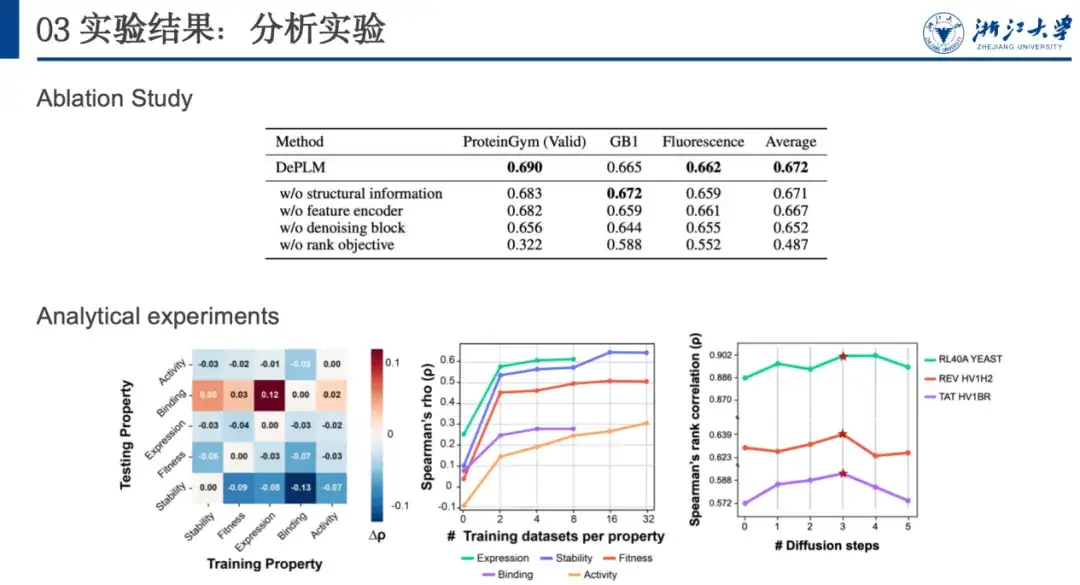
In addition, we found that the model performance improved after training on other datasets and testing on the Binding dataset. This may be attributed to the limited amount of data and low data quality of the Binding dataset, which led to its insufficient generalization ability. This inspired us toWhen optimizing proteins with new properties, if there are fewer data sets related to that property, one can consider using data with related properties for denoising and training to obtain better generalization capabilities.
Continue to deepen the protein field
The guest of this live broadcast is Wang Zeyuan, a doctoral student at the Knowledge Engine Laboratory of Zhejiang University. His team, led by Professor Chen Huajun, Researcher Zhang Qiang and others, is committed to academic research in the fields of knowledge graphs, large language models, AI for Science, etc., and has published many papers at top AI conferences such as NeurIPS, ICML, ICLR, AAAI, and IJCAI.
Zhang Qiang's personal homepage:
https://person.zju.edu.cn/H124023
In the protein field, the team not only proposed advanced models such as DePLM to optimize proteins, but also worked to bridge the gap between biological sequences and human language.To this end, they proposed the InstructProtein model.Using knowledge instructions to align protein language with human language, we explore the bidirectional generation capabilities between protein language and human language, integrate biological sequences into large language models, and effectively bridge the gap between the two languages. Experiments on a large number of bidirectional protein-text generation tasks show that InstructProtein outperforms existing state-of-the-art LLMs.
Click to see more details: Selected for ACL2024 Main Conference | InstructProtein: Aligning protein language with human language using knowledge instructions
In addition, the team also proposed a multi-purpose protein sequence design method PROPEND based on the "pre-training and prompting" framework.Through prompts for skeletons, blueprints, functional labels and their combinations, multiple properties can be directly controlled, and the method has broad practicality and accuracy. Among the five sequences tested in vitro, PROPEND achieved a maximum functional recovery rate of 105.2%, significantly exceeding the 50.8% of the classic design pipeline.
Original paper:
https://www.biorxiv.org/content/10.1101/2024.11.17.624051v1
At present, many of the results released by the team have been open sourced. They are also recruiting outstanding postdoctoral fellows, 100 people, R&D engineers and other full-time researchers on a long-term basis. Everyone is welcome to join~
Laboratory Github homepage:
http://github.com/zjunlp









