Command Palette
Search for a command to run...
Published in Nature Sub-journal! Central China Normal University Proposed DigFrag, Which Uses AI to Accurately Segment Molecular Fragments and Generate 44 drug/pesticide Molecules

Over the past few decades, fragment-based drug discovery (FBDD) has played an important role in new drug research and development by identifying small molecule fragments that have weak interactions with target proteins and optimizing the structural information of these fragments to develop more active lead compounds.
Although FBDD plays a key role in drug discovery and development, constructing and screening effective molecular fragment libraries has always been a major challenge in the field. Traditional FBDD methods rely on empirical intuition, which limits their ability to develop diverse structures. Fortunately, the emergence of AI provides a transformative solution to this challenge.
Recently, the team of Professor Yang Guangfu and Associate Professor Wang Fan from Central China Normal University developed a digital segmentation method called DigFrag.The method highlights key substructures by focusing locally on the molecular graph and dividing these substructures into fragments. Experimental results show that the fragments segmented by DigFrag show higher structural diversity, and the compounds generated based on these fragments are more consistent with the expected chemical properties. This shows that data generated by AI methods may be more suitable for the training and application of AI models.
The research, titled "DigFrag as a digital fragmentation method used for artificial intelligence-based drug design," has been published in the international academic journal Nature Communications Chemistry.
Research highlights:
* The study found that when DigFrag-based fragments are combined with AI models, they can effectively generate molecules with desired properties
* Through precise screening, the study ultimately identified 24 drug molecules and 20 pesticide molecules
* The team developed a user-friendly platform, MolFrag, which integrates multiple fragmentation technologies to support a wider range of molecular analysis and design work
Paper address:
https://doi.org/10.1038/s42004-024-01346-5
The open source project "awesome-ai4s" brings together more than 100 AI4S paper interpretations and provides massive data sets and tools:
https://github.com/hyperai/awesome-ai4s
Dataset: self-built database PADFrag, containing data on nearly 3,000 kinds of drugs
The modeling data set used by the research team mainly comes from the self-built database PADFrag. Specifically, the PADFrag database mainly includes the FDA-approved drug catalog in the DrugBank database, which contains 1,652 drugs, as well as the commercial pesticides listed by Alan Wood, totaling 1,259.
*PADFrag, a database built to explore the space of bioactive fragments for drug discovery
https://pubs.acs.org/doi/10.1021/acs.jcim.8b00285
To ensure the consistency and reliability of the data, the research team excluded compounds with non-standard structures. Subsequently, the entire dataset was divided into training set, validation set, and test set in a ratio of 8:1:1 to facilitate model training, evaluation, and testing.
DigFrag: A 3-step workflow to obtain fragments with greater structural diversity
DigFrag is an innovative digital segmentation method that uses a graph attention mechanism to identify and segment drug/pesticide fragments. Its core advantage is that it can obtain fragments with higher structural diversity from the perspective of machine intelligence rather than relying solely on human expertise.
In addition, the study integrated the fragments segmented by four methods, BRICS, RECAP, MacFrag and DigFrag, and integrated them into the DeepFMPO model framework to generate drug molecules and evaluate their performance on different indicators.
Finally, based on multiple molecular fragmentation technologies, the researchers developed a user-friendly platform MolFrag to support molecular segmentation work.
Specifically, the workflow of this study is divided into three parts:
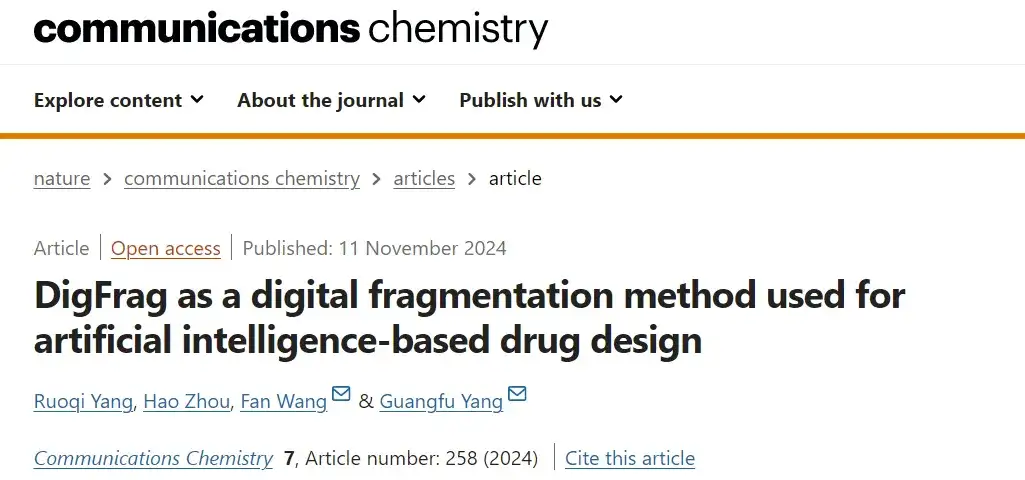
First, the AI-based fragmentation approach:This study is based on the Graph Neural Network (GNN) architecture and uses the DigFrag method to fragment molecules.
As shown in Figure A above, the researchers defined the molecular graph as G=(V, E), where V represents nodes, corresponding to atoms in molecules, and E represents connecting edges, corresponding to chemical bonds between atoms. In this process, based on the feature extraction network (feature matrix) of the graph attention mechanism, the original molecular graph is first input into a series of attention layers, with the aim of obtaining a separate embedding representation for each atom. These atomic embeddings are then aggregated to form a unified vector, also called a super node. Finally, through further attention layer processing, the embedding representation of the entire fragment is obtained.
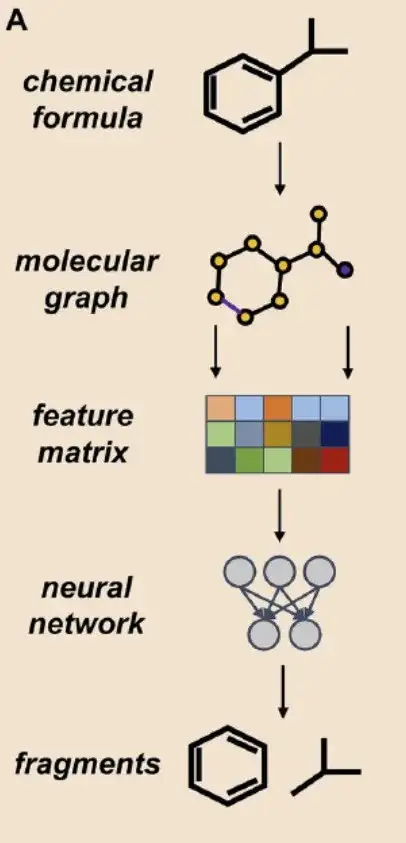
Second, the Actor-Critic model frameworkAs shown in Figure B below, in order to further clarify the impact of digital segmentation on fragment-based deep generative models, the researchers integrated fragments segmented by four methods: BRICS, RECAP, MacFrag and DigFrag, and used an open source fragment-based reinforcement learning two-dimensional molecule generation tool DeepFMPO architecture for research.
*DeepFMPO is an Actor-Critic reinforcement learning model that obtains the desired compound by replacing fragments in the compound.
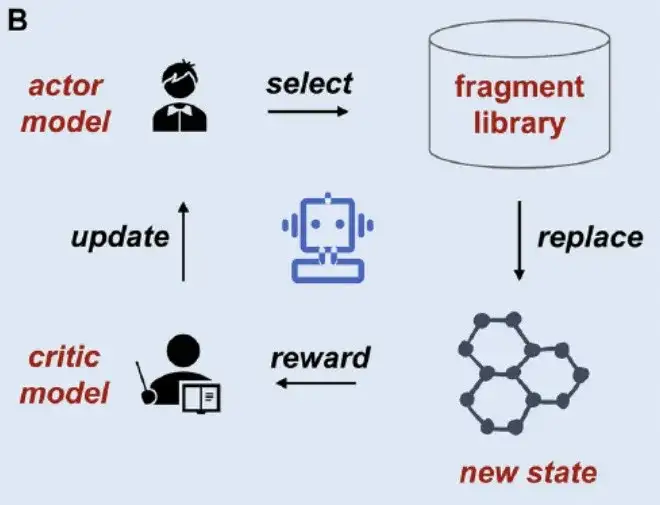
Third, establish an online platform:Although there are many molecular fragmentation methods, there is a lack of easy-to-use online servers. Therefore, as shown in Figure C above, this study developed a user-friendly platform MolFrag based on various fragmentation technologies. The platform seamlessly combines four molecular fragmentation methods: BRICS, RECAP, MacFrag, and DigFrag, ensuring that researchers of different professional levels can use it.
MolFrag platform address:
https://dpai.ccnu.edu.cn/MolFrag
Research results: DigFrag segmented molecular fragments have higher diversity
DigFrag fragments have a large number of rotatable bonds
The study first trained the model to accurately segment drug and pesticide fragments. Then, the researchers conducted a five-fold cross-validation to compare the model accuracy, area under the curve (AUC), and Matthews correlation coefficient (MCC) of fragments obtained by DigFrag with those obtained by traditional (RECAP, BRICS) and the latest (MacFrag) methods. As shown in the table below, in terms of the distribution of the properties of drug fragments, the fragments segmented by DigFrag are more similar to those segmented by BRICS.
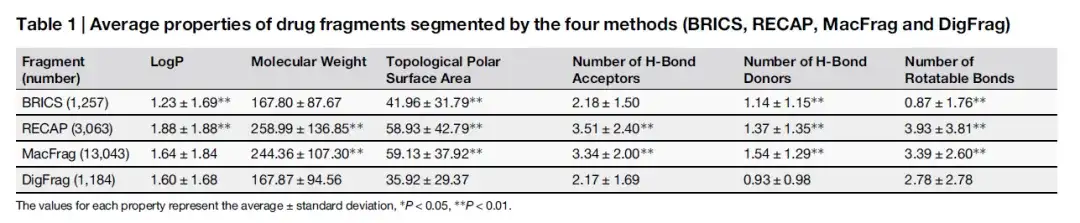
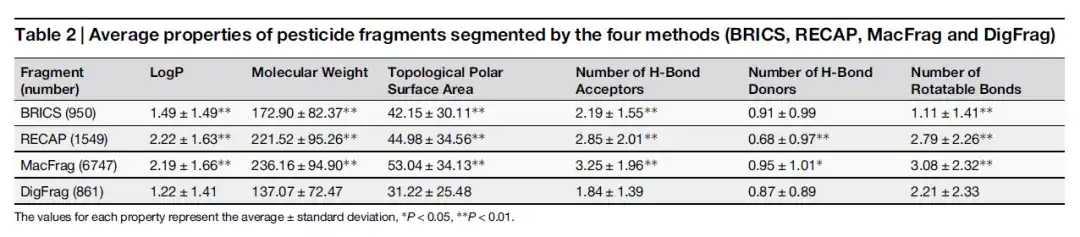
As shown in the table below, although the molecular weight and number of H-Bond Acceptors of drug fragments separated by DigFrag are similar to those separated by BRICS, the number of rotatable bonds is higher, which may be related to its unique ring structure breaking mode. In terms of pesticide fragments, the average molecular weight of fragments separated by DigFrag is lower.
DigFrag segmented fragments have higher structural diversity
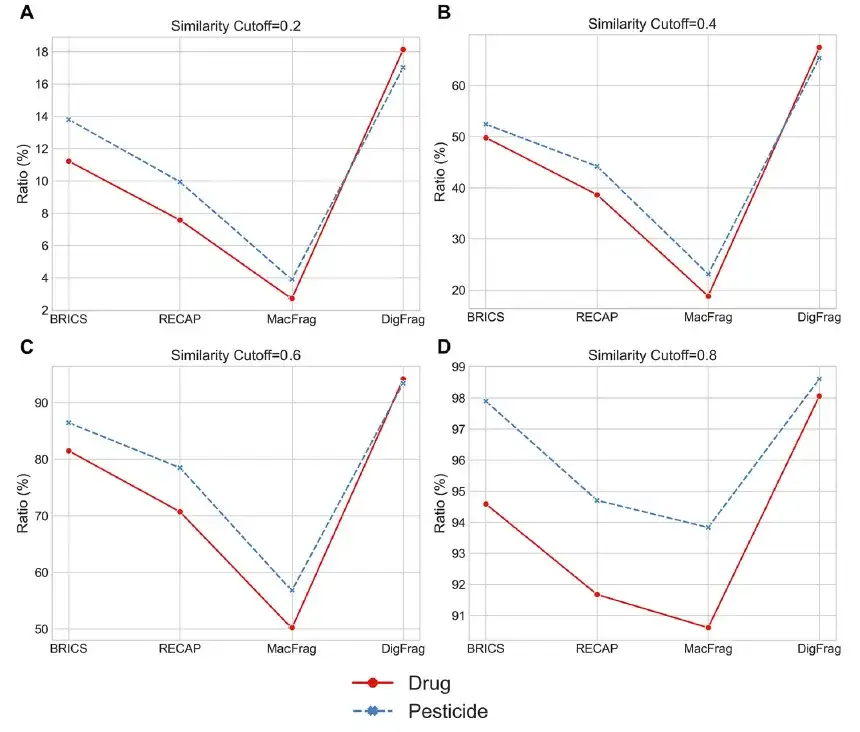
The focus of this study was to evaluate the structural diversity of segmented fragments when comparing the DigFrag method with traditional methods (RECAP and BRICS) and the latest method (MacFrag). The results showed that the fragments segmented by DigFrag in drug and pesticide fragments had a lower repetition rate than the other three methods, 9.97%-21.37% and 8.94%-15.20%, respectively, indicating that it can generate unique fragments. MacFrag covered most of the fragments of BRICS and RECAP, suggesting that it is not completely innovative, but an extension of traditional methods.

The researchers also used the t-SNE algorithm to visualize the chemical space distribution. As shown in the figure below, DigFrag performed well in the fragment clustering ratio, especially when the similarity thresholds were at 0.4 and 0.6, showing higher structural diversity.
DigFrag-based models generate higher quality molecules
On the MOSES benchmark platform, the study compared the performance of different generative models. The data in the two tables below show that the DigFrag-based model achieved a Filters score of 0.828, showing higher safety, which may be attributed to the comprehensive consideration of toxicity and stability in the fragmentation process of deep learning.
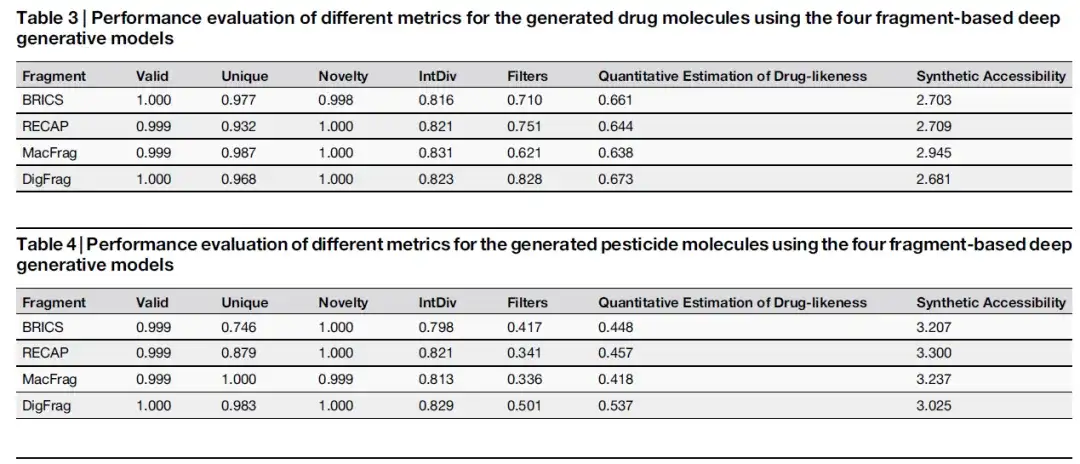
As shown in the figure below, in terms of pesticide molecules, the molecular fragments generated by the DigFrag-based model performed well in terms of SMILES validity, novelty, skeleton diversity, and structure alerts. In addition, the drug and pesticide molecular fragments generated by the DigFrag model outperformed other models in the average value analysis of quantitative estimation (QED) and synthetic accessibility (SA).
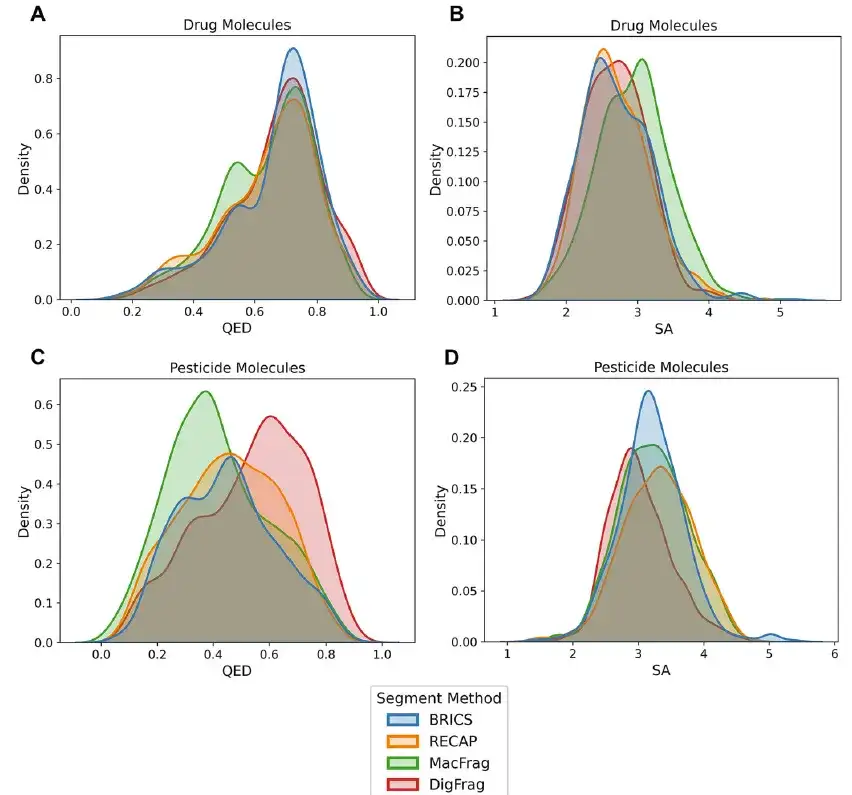
In addition, the molecular fragments segmented by DigFrag have the highest similarity with the MOSES dataset in terms of molecular weight, QED and SA property distribution. These results show that the DigFrag-based model can produce higher quality molecules, while emphasizing the preference of AI models for AI-derived data in molecular design, highlighting the application advantages of AI technology in this field.
Selected 44 high-efficiency and low-energy drug and pesticide molecules
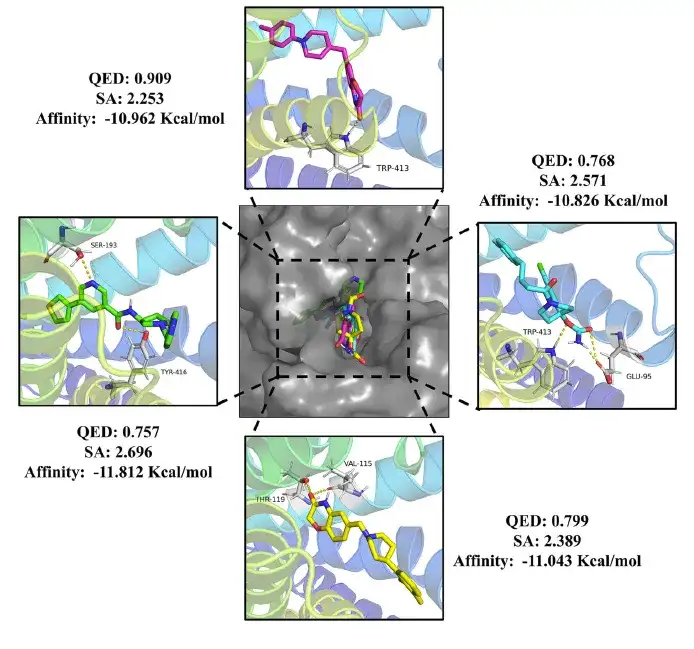
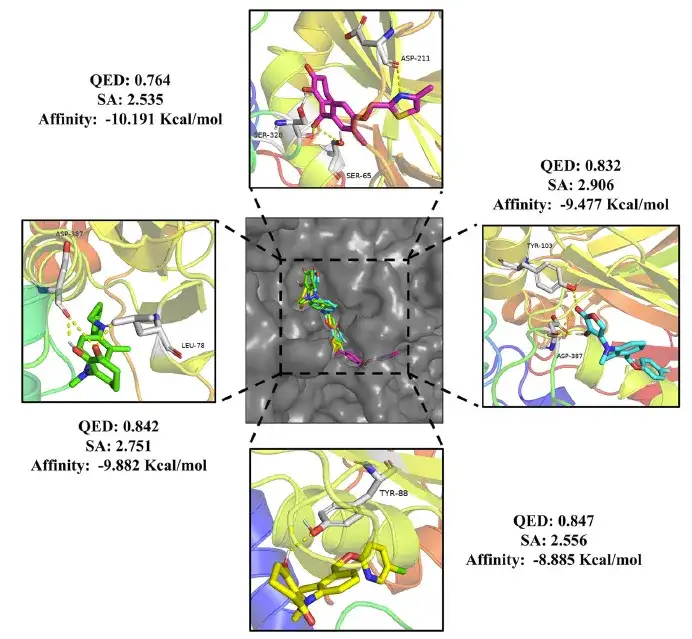
Finally, after precise screening, the study identified 24 drug molecules and 20 pesticide molecules, all of which met the criteria of QED values greater than 0.75, SA values less than 3, and binding free energy lower than domperidone (-10.7 Kcal/mol) and methotriazine (-8.4 Kcal/mol).
The study further analyzed the interaction between these molecules and the target. As shown in the figure below, the study found that the drug molecules can effectively bind to the DRD2 active pocket and form hydrogen bonds with key amino acid residues.
Moreover, as shown in the figure below, the pesticide molecules form hydrogen bonds with the amino acid residues of HPPD to stabilize the binding. Compared with the positive drugs, the generated compounds also show different binding modes, suggesting that there may be different pharmacological mechanisms, which provides new directions for future research.
The application of AI in drug research reshapes the rules of the game
At present, the application of AI in drug research is becoming more and more in-depth. Through deep learning networks, AI models can analyze complex biological data and chemical structures to predict the activity and selectivity of drug molecules.
The team of Professor Yang Guangfu and Associate Professor Wang Fan mentioned in this study also jointly developed a multimodal deep learning architecture model Pesti-DGI-Net for predicting pesticide-like properties earlier this year. It can predict the pesticide-like properties of compounds by integrating three molecular representation forms: molecular descriptors, molecular images, and molecular graphs. The results show that Pesti-DGI-Net has excellent performance in multiple indicators.
Paper link:
https://doi.org/10.1016/j.compag.2024.108660
In addition, AI has recently achieved fruitful results in the field of drug properties research. Not long ago, the Shanghai Institute of Nutrition and Health of the Chinese Academy of Sciences built a dual-view deep learning model JointSyn to predict the synergistic effect of drug combinations. The results show that JointSyn outperforms existing state-of-the-art methods in terms of prediction accuracy and robustness on various benchmarks.
Paper link:
https://doi.org/10.1093/bioinformatics/btae604
In addition to its application in drug property prediction, AI technology has also achieved remarkable research results in drug design optimization, toxicology and safety assessment, clinical trial design, and patient selection. It is foreseeable that the application of AI in drug property research is reshaping the rules of the game for drug development. With the continuous advancement of technology, it may bring safer and more effective treatment options to patients by improving the accuracy of predictions, optimizing drug design, and reducing development costs and time.








