Command Palette
Search for a command to run...
Selected for NeurIPS 2024! Westlake University Proposed the Universal Molecular Inverse Folding Model UniIF, Which Further Complements AlphaFold 3

Molecular inverse folding plays a key role in drug and material design, allowing scientists to synthesize new molecules with ideal structures. Past research has mostly focused on the inverse folding of large or small molecules, but little attention has been paid to the inverse folding of general molecules.
There are three major challenges in building a unified general model:① Unit differences: Large molecules generally use predefined microstructures as basic units, such as amino acids for proteins and nucleotides for RNA; small molecules use atoms as basic units; ② Geometric feature extraction: Different studies use a variety of strategies for geometric feature extraction, such as distance, angle, and tensor product, and lack a unified characterization method; ③ System scale: Small molecules allow global attention mechanisms to learn long-term dependencies, but this often does not work on large molecules.
To address the above challenges and further complement the progress made by RoseTTAFold All-Atom and AlphaFold 3 in molecular structure prediction,A team from the Future Industry Research Center of Westlake University proposed a unified model, UniIF, for the inverse folding of all molecules.The researchers conducted comprehensive experiments on multiple tasks such as protein design, RNA design, and material design to demonstrate the effectiveness of UniIF. The results showed that UniIF achieved state-of-the-art performance on all tasks.
The related research, titled "UniIF: Unified Molecule Inverse Folding", was selected for the top conference NeurIPS 2024.
Research highlights:
* The proposed unified model UniIF provides a versatile and effective solution for general molecular inverse folding
* The model is unified at two levels: at the data level, a unified block graph data form for all molecules is proposed, including the construction of the local coordinate system and the initialization of geometric features; at the model level, a geometric block attention network is introduced to capture the three-dimensional interactions of all molecules
* The researchers demonstrated that the proposed method outperforms state-of-the-art methods in three major tasks: protein design, RNA design, and material design. This achievement may have a positive impact on the machine learning, drug discovery, and materials science communities.
Paper address:
https://arxiv.org/abs/2405.18968
Follow the official account and reply "Molecular Reverse Folding" to get the complete PDF
The open source project "awesome-ai4s" brings together more than 100 AI4S paper interpretations and provides massive data sets and tools:
https://github.com/hyperai/awesome-ai4s
Dataset: Select the corresponding dataset for three task experiments
In the protein design task,The researchers evaluated UniIF on the CATH4.3 dataset, which is split according to the CATH topological classification code, resulting in 16,631 training samples, 1,516 validation samples, and 1,864 test samples.
To evaluate the generalization ability, the researchers adopted a time-partitioning strategy, considering that some baselines use pre-trained ESM2 models, which poses a risk of data leakage. The time-partitioning evaluation assigns data before a specific date to the training set, and data after that date to the test set. For the time-partitioning evaluation of structures, the CASP15 dataset is used, which contains new crystal structures not seen during training; for the time-partitioning evaluation of sequences, the NovelPro dataset is used, which contains 76 protein sequences published within 30 days before November 23, 2023, and the structures are predicted by AlphaFold 2.
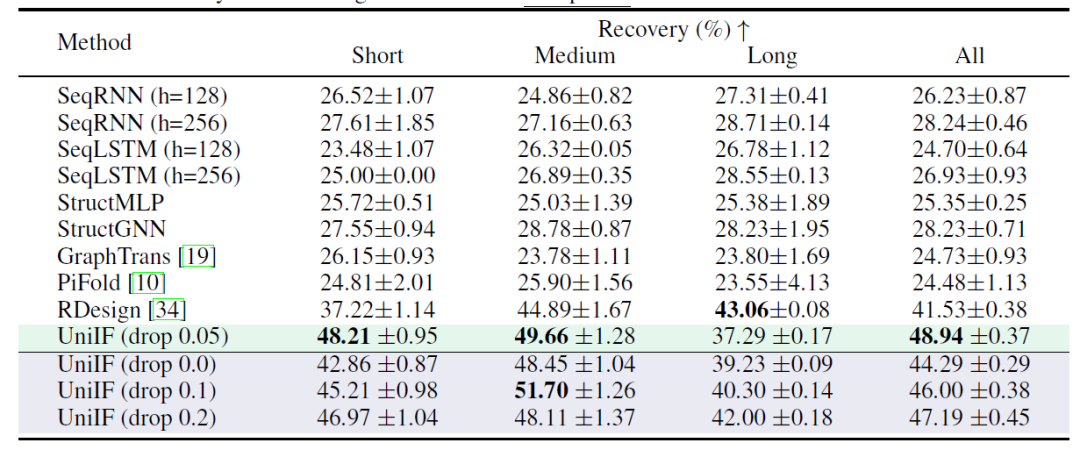
In RNA design tasks,The researchers conducted RNA experiments on a dataset collected by RDesign, which contains 2,218 RNA tertiary structures, which are divided into a training set (1,774 structures), a test set (223 structures), and a validation set (221 structures) according to their structural similarity. Due to the small number of data samples, the researchers reported the median recovery rate and its standard deviation of 3 independent runs.
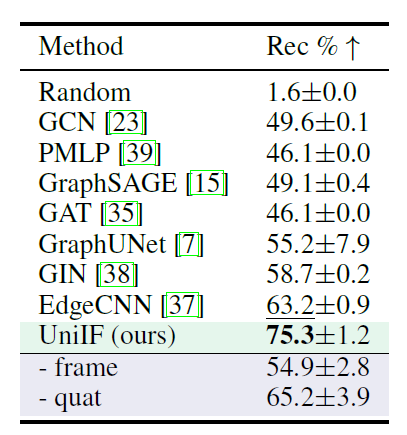
In the material design task,The researchers evaluated UniIF on the CHILI-3K dataset, which consists of graphs of nanomaterials derived from single metal oxides. The dataset includes 53 metal elements and one non-metal element (oxygen), totaling 3,180 graphs, 6,959,085 nodes, and 49,624,440 edges.
Model architecture: UniIF, a unified model for general molecular inverse folding
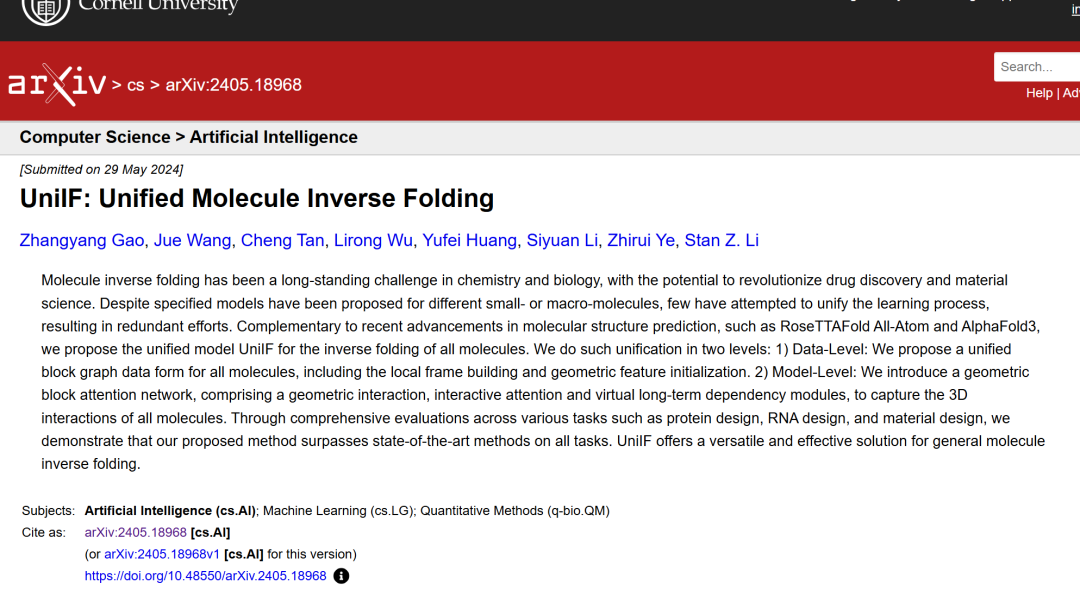
As shown in the figure below, the researchers proposed a unified model for general molecular inverse folding.
① The model converts all types of molecules into block graphs - for macromolecules, a predefined framework based on amino acids and nucleotides is used; for small molecules, a local framework for each block is learned through a layer of GNN;
② Use the Geometric Featurizer to initialize the geometric node features and edge features;
③ A Block Graph Attention layer is proposed, based on which a Block Graph Neural Network is constructed to learn to express rich block representations;
④ Finally, we demonstrate that UniIF can achieve competitive results in a variety of tasks, including protein design, RNA design, and material design.
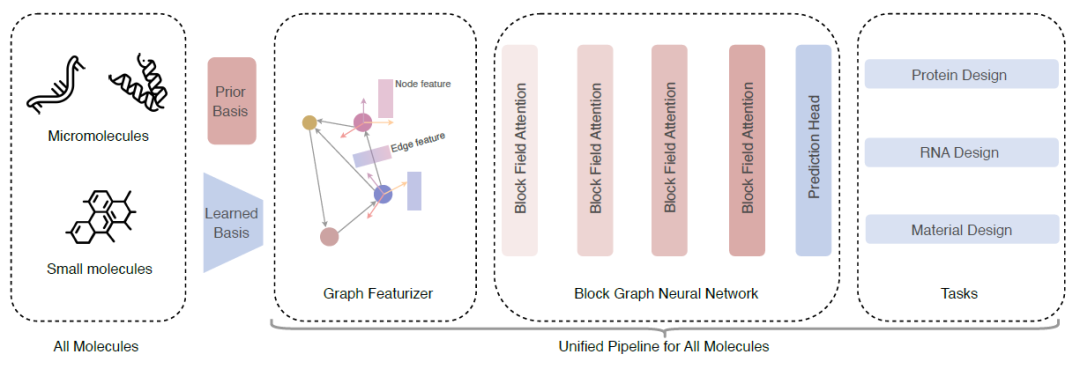
Building Block Diagram:The first step of the model architecture is to introduce block graphs to represent all types of molecules. The key is to convert irregular sets of atoms (of varying sizes) into regular block representations (fixed size). The researchers introduced a frame-based block representation to unify the modeling of all molecules. A block contains an equivariant frame and an invariant feature vector, and a local frame contains an axis matrix and a displacement vector. For large molecules, the axis matrix is predefined based on amino acids and nucleotides; for small molecules, because small molecules have no prior common structural patterns, the axis matrix needs to be learned. Given a molecule containing n blocks, the researchers used the kNN algorithm to construct a block graph.
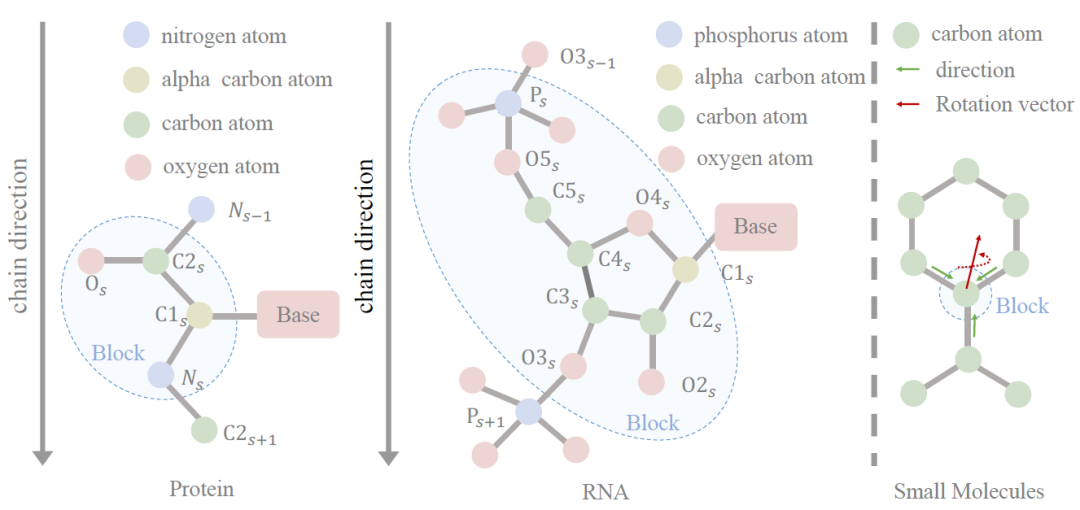
Block graph feature extraction:For small molecules, predefined local frames are not available, so researchers need to learn a local frame for each atom - that is, given a molecule, a layer of GNN is used to initialize the atomic representation, and then a geometric feature extractor is used to initialize the geometric node features and edge features.
Block Graph Attention Module:The researchers introduced a geometric block attention network, including geometric interaction, interaction attention, and virtual long-term dependency modules, to capture the three-dimensional interactions of all molecules.
Research results: UniIF outperforms state-of-the-art methods on all tasks
The researchers demonstrated the effectiveness of UniIF through multiple inverse folding tasks and ablation studies, including:
* Protein design (T1): Designing protein sequences that can fold into target structures
* RNA Design (T2): Design RNA sequences that can fold into target structures
* Material Design (T3): Discovering stable compositions from known material structures
① Protein design (T1)
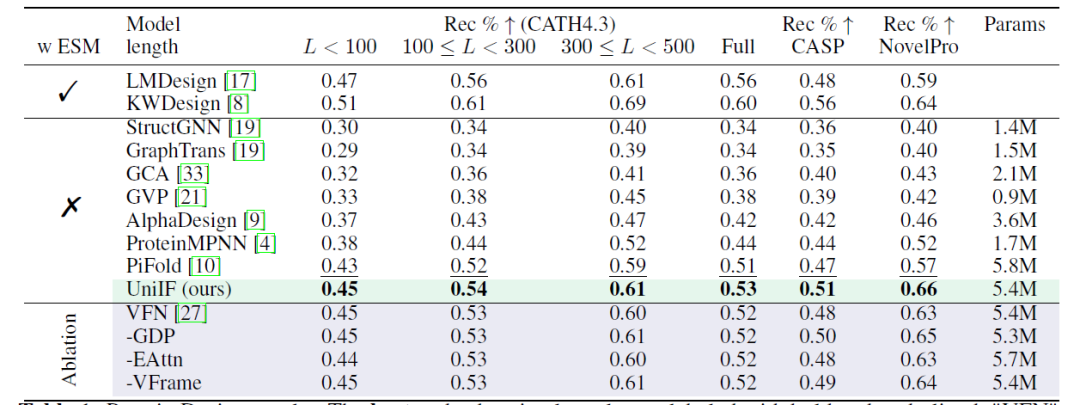
Protein design aims to design protein sequences that can fold into target structures, and researchers provided results under different settings (with and without ESM2) and multiple datasets (CATH4.3, CASP, NovelPro). As shown in the following table: Using a pure inverse folding model without ESM2, UniIF achieved the best performance on all datasets, demonstrating its effectiveness.
*LMDesign and KWDesign include ESM2; StructGNN, GraphTrans, GCA, GVP, AlphaDesign, ProteinMPNN, and PiFold do not include ESM2
On CATH4.3, the overall improvement is limited due to the strong baseline model, but the time partitioning evaluation highlights the advantage of UniIF in generalization ability. UniIF surpasses the strong baseline PiFold with fewer learnable parameters. In the time partitioning evaluation, UniIF surpasses all baselines, including the ESM2-based method, by a significant margin. On NovelPro, which contains new sequences, UniIF outperforms LMDesign and KWDesign, which use ESM2 for sequence optimization.This indicates that UniIF has superior generalization ability, which is crucial for practical applications.
② RNA design (T2)
The goal of RNA design is to design RNA sequences that can fold into a target structure. As shown in the table below, UniIF achieved the best performance in all cases, which is a significant improvement because previous strong baseline models such as PiFold only performed well in protein design.UniIF is the first model to achieve state-of-the-art performance in both protein and RNA design tasks, demonstrating its versatility and effectiveness.
③ Material Design (T3)
Finding stable atomic combinations from known material structures is crucial for the discovery of new materials, so the researchers also evaluated UniIF's performance on this new task. As shown in the table below,UniIF significantly outperforms all baseline models.
④ Case Study
In the figure below, the researchers show the designed protein and RNA sequences. In addition, they use AlphaFold 3 to refold the designed sequences into structures - the real structure (gray), PiFold structure (green) and UniIF structure (pink) are aligned and compared. The researchers observed thatUniIF achieves improvements in both recovery rate and root mean square deviation (RMSD), demonstrating its effectiveness in the inverse folding task.

UniIF model further complements AlphaFold 3
General molecular learning has received increasing attention in recent years, and RoseTTAFold All-Atom (RFAA) and AlphaFold 3 are two representative models that have achieved remarkable success in this direction.
On March 7, 2024, David Baker published a research paper titled "Generalized biomolecular modeling and design with RoseTTAFold All-Atom" in Science. The team developed RoseTTAFold All-Atom (RFAA), which combines residue-based representations of amino acids and DNA bases with atomic representations of all other groups to model covalently modified components including proteins, nucleic acids, small molecules, metals, and given sequences and chemical structures.
Original paper:
https://www.science.org/doi/10.1126/science.adl2528
On May 9, 2024, Demis Hassabis, John Jumpe and others published a research paper titled "Accurate structure prediction of biomolecular interactions with AlphaFold 3" in Nature. The study launched AlphaFold 3, a new model that can predict the structure of complexes containing almost all molecular types in the Protein Data Bank, including how ligands (small molecules), proteins, and nucleic acids (DNA and RNA) come together and interact, as well as predicting the structural effects of post-translational modifications and ions on these molecular systems, thereby helping researchers to accurately observe the structure of biomolecular systems at the atomic level.
Original paper:
https://www.nature.com/articles/s41586-024-07487-w
Looking at the two models in detail, RFAA uses atom-bond diagrams to represent small molecules and frame diagrams to represent large molecules; AlphaFold 3 uses a two-layer representation, namely atom representation and tag representation, which is applicable to all molecules. The tag concept is equivalent to the block concept mentioned above, representing a group of atoms, such as amino acids or nucleotides.
GET and EPT are two recently proposed models that use block representations for both small and large molecules and introduce new isovariant transformer frameworks. Unlike RFAA, which specifies atom-bond diagrams for small molecules, the UniIF model introduced in this paper uses a unified block diagram for all molecule types, does not require atom-bond diagrams, and also introduces a vector basis for each block, which is different from AlphaFold 3, GET, and EPT.
Since the challenge of building a universal molecular model has been solved to some extent,The UniIF model can be seen as a further supplement to the progress made in molecular structure prediction by "predecessors" such as RoseTTAFold All-Atom and AlphaFold 3.In the future, continuously iterating biological big models will help researchers re-understand the biological world and rethink drug discovery, thus benefiting all mankind.
References:
1.https://arxiv.org/abs/2405.18968
2.https://mp.weixin.qq.com/s/8OvxVlUuZZZ2gcepIl5UBw
3.https://www.jiqizhixin.com/articles/2024-03-08-6
4.https://m.thepaper.cn/newsDetail_forward_28984037









