Command Palette
Search for a command to run...
Published in Nature Sub-journal! The First Author of the Paper Explains the Small Sample Learning Method of Protein Language Model to Solve the Problem of Lack of Wet Experimental Data

In the third episode of the "Meet AI4S" series, we have the honor of inviting Zhou Ziyi, a postdoctoral fellow at the Institute of Natural Sciences of Shanghai Jiao Tong University and the Shanghai National Center for Applied Mathematics,His research group at Shanghai Jiao Tong University, Hong Liang, focuses on AI protein and drug design and molecular biophysics. The research group has achieved fruitful results, publishing 77 research papers so far, many of which have topped Nature journals.
In this sharing session, Dr. Zhou Ziyi shared the team’s latest research results under the title “Small Sample Learning Method for Protein Language Model” and explored new ideas for AI-assisted directed evolution.
Research Background of Protein Language Model (PLM)
Protein & Protein Engineering
Protein is the main carrier of biological functions and the executor of life activities. The natural amino acid ammonia undergoes a dehydration condensation reaction to form a protein residue sequence, which is then folded into a tertiary structure. Changing the type of amino acids in a protein will affect its structure and function.
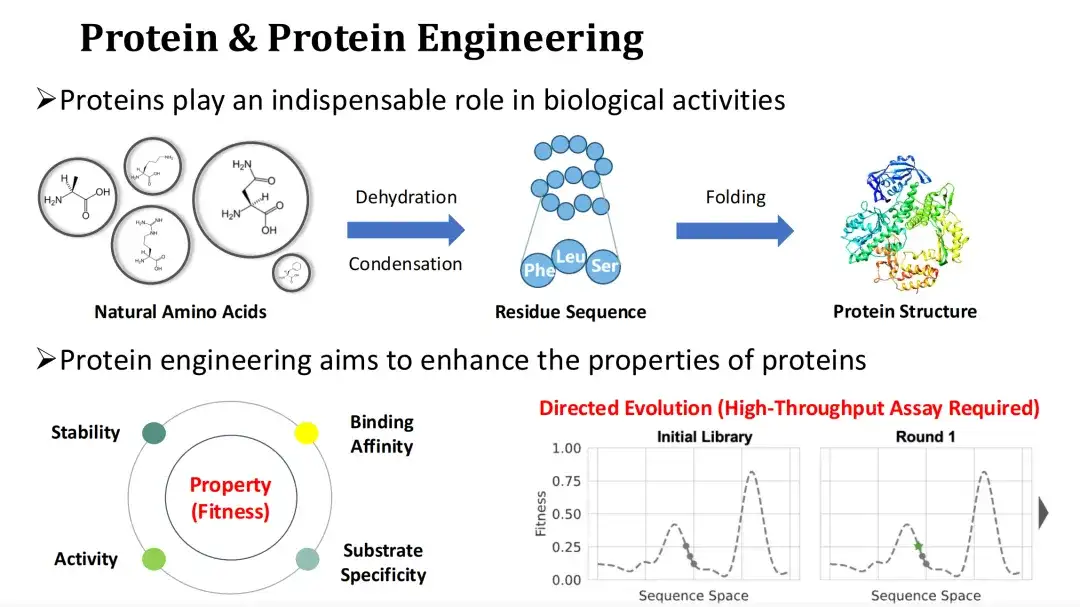
Since natural proteins are often difficult to meet industrial or medical needs, protein engineering hopes to improve the functional properties of proteins, such as catalytic activity, stability, binding ability, etc., by mutating them.
We usually refer to the quantification of protein functional properties as Fitness. Directed evolution is now the mainstream protein engineering method.It relies on random mutation and high-throughput experiments to find high-Fitness mutants, but the experimental cost is high.The topic I will share today is how to use AI methods to predict Fitness and thus reduce experimental costs.
PLM Architecture
We know that language models represented by ChatGPT are very powerful and can perform high-quality text understanding and generation. These language models are pre-trained on massive amounts of text to learn the statistical laws of text, master basic grammar, and the semantics of words in context. So, can protein language models be trained similarly on massive amounts of protein sequences? The answer is yes.

The protein language model (PLM) has three main functions. First, PLM can model the co-evolutionary information of protein sequences and learn the interdependencies and evolutionary constraints between residues.Just like a natural language LM can learn the grammar of a text, a PLM can use this ability to estimate which mutations are harmful or beneficial, and thus predict the fitness of the mutation.
Secondly, in addition to Fitness prediction, PLM can also calculate the vector representation of proteins.These representations can be used for structure prediction or protein mining, and after fine-tuning, can also perform function prediction.
Finally, PLM can perform conditional protein generation like ChatGPT to achieve de novo protein design.
The architecture of PLM is similar to that of natural language LM, which is divided into autoregressive model and masked model.The network structure of these two models both uses Transformer, which consists of a self-attention mechanism and a fully connected layer. The main difference lies in the pre-training objectives.
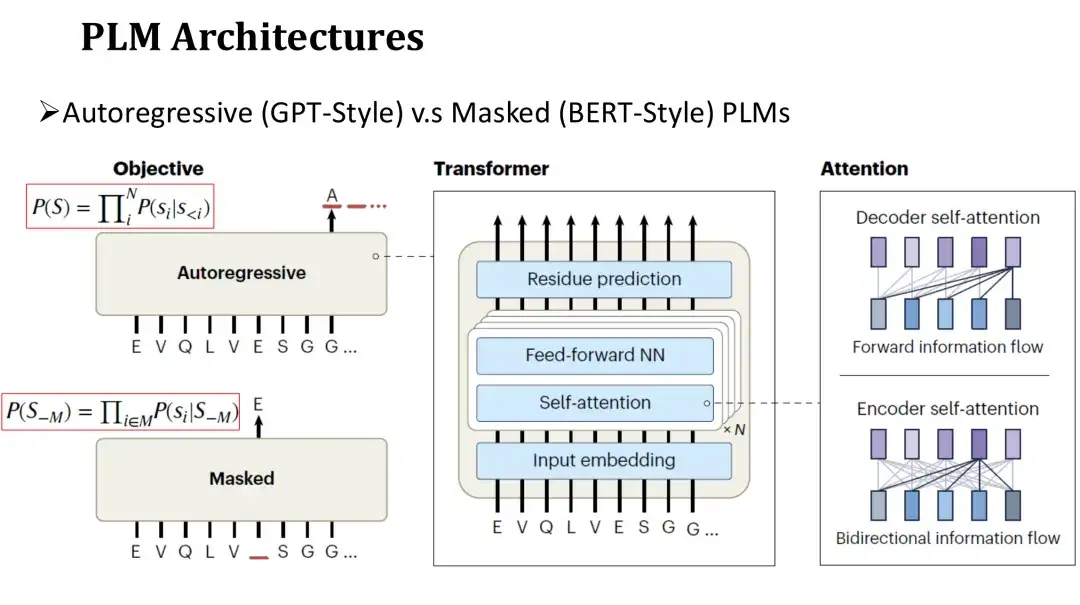
The pre-training goal of the autoregressive model is to generate the next amino acid in sequence from left to right.The goal of the masking model is to restore the randomly masked amino acids, similar to filling in the blanks. Since the autoregressive model can only rely on the generated sequence on the left when predicting each amino acid, its attention is unidirectional.The masking model can see the amino acids on both sides of the masked position during prediction.Therefore, its attention is two-way.
Two hot research directions of PLM
At present, the research hotspots of PLM are mainly divided into two directions. The first is retrieval-augmented PLM.During training or prediction, this type of model takes the multiple sequence alignment (MSA) of the current protein as an additional input, and improves the prediction performance through the retrieved information. For example, MSA Transformer and Tranception are typical models of this type.
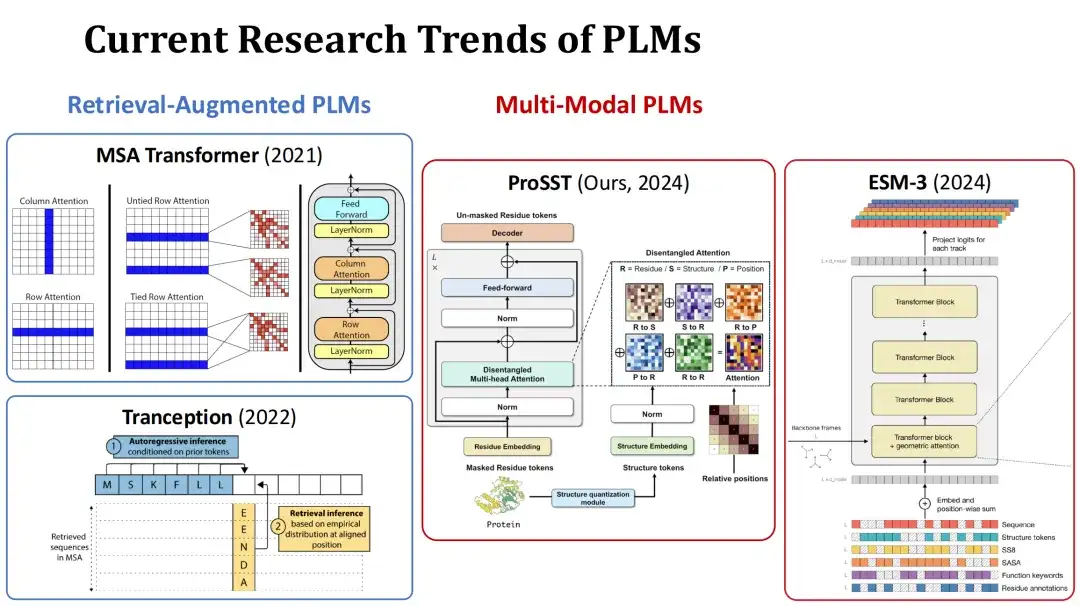
The second is Multi-Modal PLM.In addition to protein sequences, this type of model also takes protein structure or other information as additional input to enhance the model's representational capabilities. For example, the ProSST model submitted by our group this year quantifies protein structure into a structure token sequence and inputs it into the Transformer model together with the amino acid sequence, fusing these two types of information through a separate attention mechanism. Another example is the ESM-3 model of the same period, which considers richer information, including amino acid type, complete tertiary structure, tertiary structure token, secondary structure, solvent accessible surface area (SASA), and functional descriptions of proteins and residues, a total of 7 inputs.
Unsupervised and Supervised Fitness Prediction
Next, we will discuss the Fitness prediction problem.Since PLM can model the probability distribution of protein sequences, it can be directly used for fitness prediction of mutations without labeled data. This method is called zero-shot prediction or unsupervised prediction.

Specifically, PLM scores mutations by calculating the log-likelihood ratio between mutants and wild-type. For autoregressive models, the probability of a sequence, P, is the product of the probabilities of generating each amino acid. The score of a mutation can be obtained by subtracting the logP of the wild-type from the logP of the mutant. Intuitively, it is to compare how much higher the probability of a mutation is relative to the wild-type, and then evaluate the impact of the mutation. This is an empirical evaluation method.
For the masking model, it is impossible to directly calculate the probability of the entire sequence, but it can first mask a certain point and then estimate the probability distribution of the amino acid at this point. Therefore, for each mutation position, the logP of the predicted mutant amino acid after masking can be subtracted from the logP of the wild-type amino acid, and then the difference of all positions can be added to obtain the score of the mutant.
In addition, since PLMs provide vector representations of protein sequences, they can also be fine-tuned to achieve supervised Fitness prediction when there is sufficient experimental data.
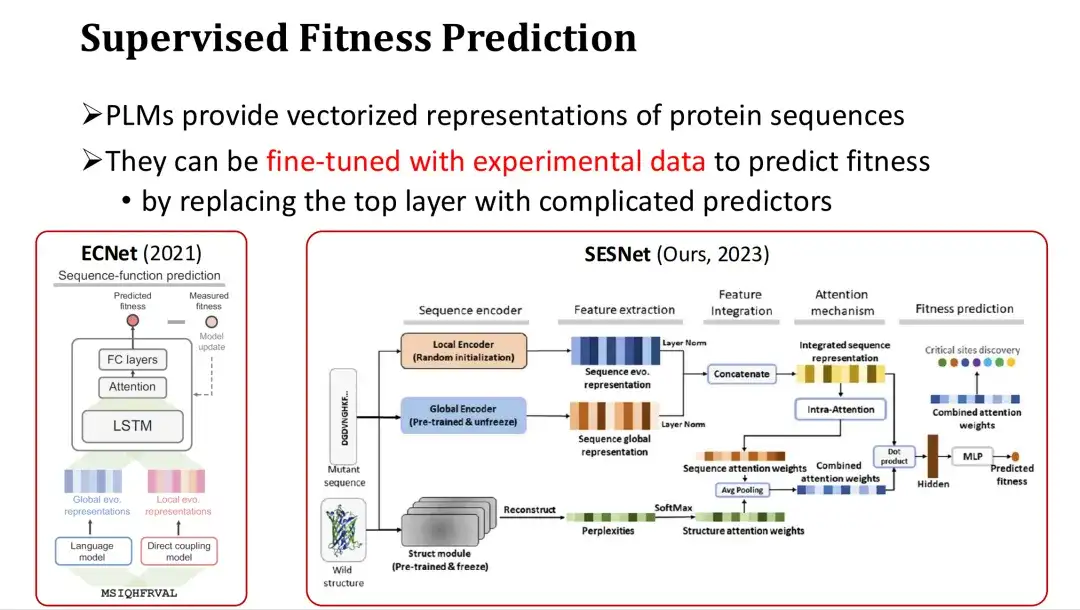
The specific approach is to add an output layer for predicting Fitness (such as an attention mechanism or a multi-layer perceptron MLP) after the last layer of PLM features, and use Fitness Label for full or partial training. For example, ECNet adds MSA features based on the features of the large model, integrates them through LSTM, and performs supervised training. The SESNet model developed by our research group last year integrates the sequence features of ESM-1b, the structural features of ESM-IF, and the MSA features for supervised Fitness prediction.
Introduction to FSFP Method: A Small Sample Learning Method for PLM
Importance of small sample learning for Fitness prediction
Before introducing the FSFP method, we need to first clarify the importance of small sample learning in Fitness prediction. Although unsupervised methods do not require labeled data for training, their zero-shot scoring accuracy is low. In addition, because the scores based on the log-likelihood ratio can only reflect certain natural laws of proteins, they are also difficult to effectively predict the non-natural properties of proteins.

On the other hand, although supervised learning methods are very accurate, due to the huge number of PLM parameters, they require large-scale experimental data to train in order to significantly improve performance. The evaluation of supervised learning models generally involves splitting existing high-throughput datasets 8:2, and the training set of 80% may already contain tens of thousands of data, which is very expensive to obtain in practice.
To address this problem, we proposed the FSFP method, which is a small sample learning method suitable for PLM. This method can significantly improve the Fitness prediction performance of PLM using a small number of training samples (dozens). At the same time, the FSFP method has strong flexibility and can be applied to different PLMs.
FSFP method: ranking learning for Fitness
Previous supervised learning methods have all treated Fitness prediction as a regression problem, that is, optimizing the model by calculating the mean square error (MSE) between the model output and the Fitness Label. However, under small sample conditions, regression models are very prone to overfitting, and the training loss drops very quickly. Therefore, we changed our thinking and did not do regression, but ranking learning, which only requires accurate sorting, not exact fitting values.
This approach has two major advantages. First, the ranking itself meets the basic needs of protein engineering, which only requires measuring the relative effectiveness of mutations. Second, the ranking task is simpler than predicting absolute values.
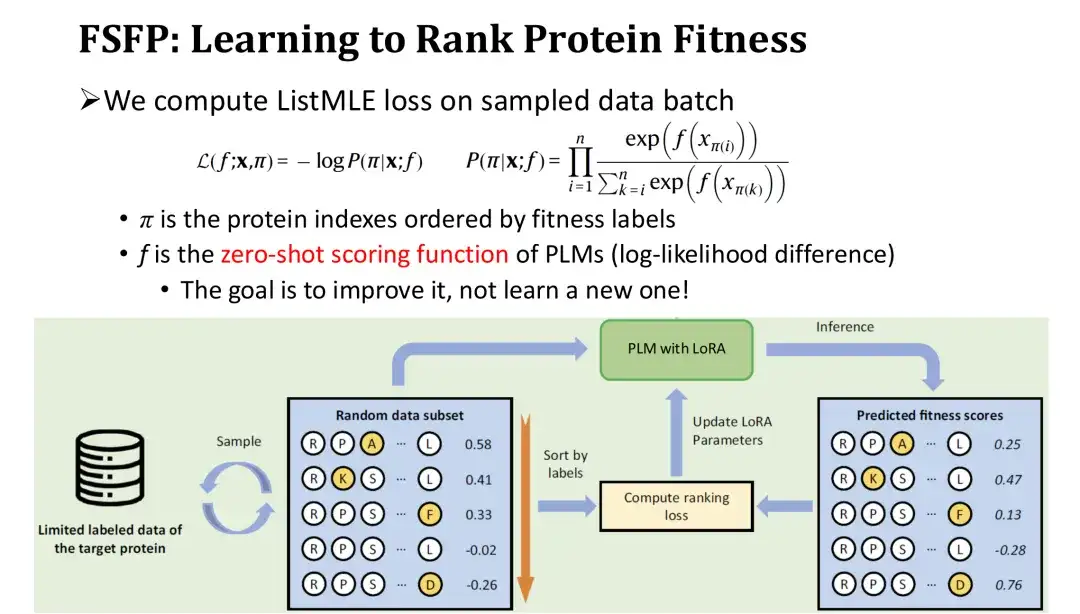
In the training iteration, we sort the sampled set of mutants in reverse order according to their labels, and then calculate the ranking loss - ListMLE based on the model's prediction values for these mutants.The closer the ranking of the model's predicted values is to the true ranking, the smaller the loss. Among them, we use the zero-shot scoring function based on the log-likelihood ratio as the model's scoring function for mutations f. The purpose of this is to use the zero-shot score as a starting point and gradually correct it with training data to improve performance without reinitializing a module, thereby reducing the difficulty of training.
FSFP Method: Parameter-Efficient Fine-Tuning of PLM
Since the number of parameters in a PLM is usually as high as hundreds of millions, fine-tuning the entire model with very little data will inevitably lead to overfitting.Therefore, we introduced a second technique, LoRA, to limit the number of trainable parameters of the model.
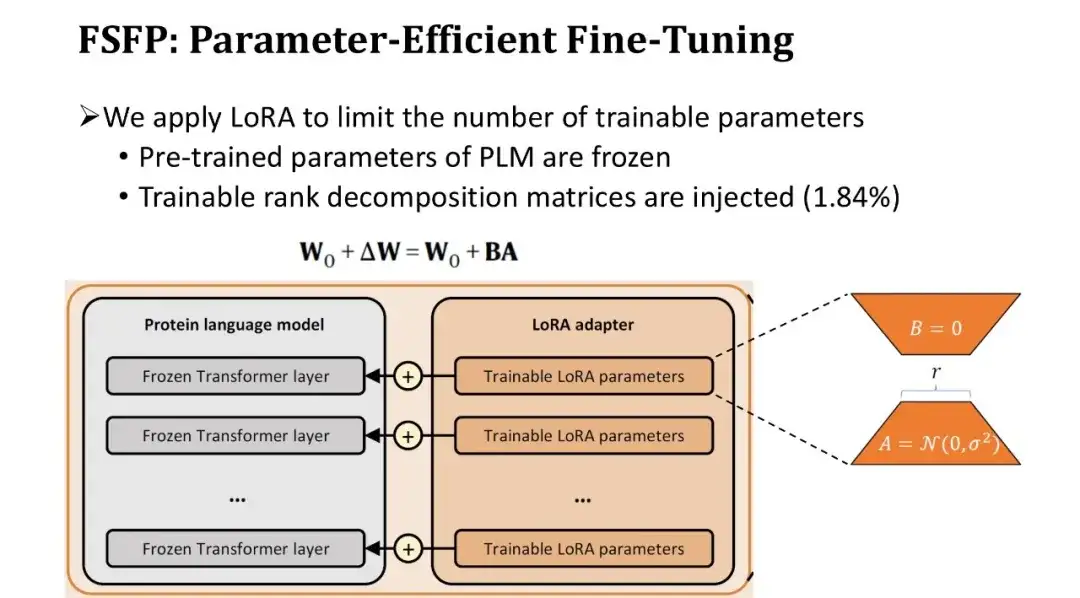
LoRA inserts a pair of trainable rank decomposition matrices in the fully connected layer of each Transformer block, keeping the pre-trained parameters unchanged. Because the rank decomposition matrix is very small, the number of trainable parameters can be reduced to the original 1.84%. Although the number of trainable parameters is reduced, the learning ability of the model can still be guaranteed because each layer of the Transformer is fine-tuned.
FSFP Method: Applying Meta-Learning to Fitness Prediction
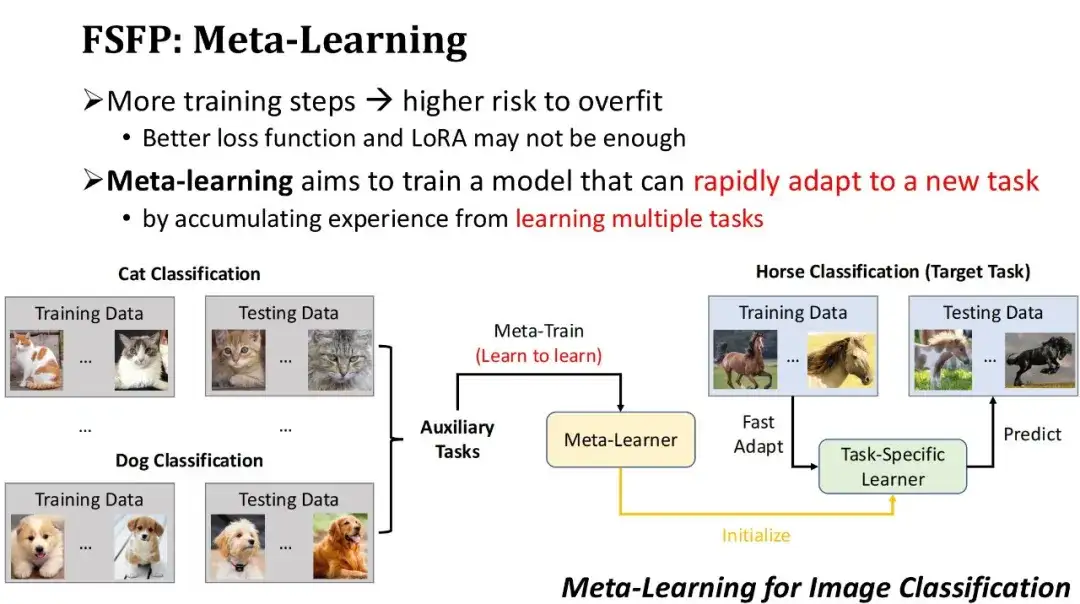
To avoid overfitting, we not only use a better loss function, but also limit the number of trainable parameters through LoRA technology. However, even so, if there are too many training iterations on small sample training data, there is still a risk of overfitting. Therefore, we hope to quickly improve model performance with fewer training iterations.Based on this requirement, we adopted the third technology - meta-learning. The basic idea of meta-learning is to first let the model accumulate experience on some auxiliary tasks to obtain an initial model, and then use the initial model to quickly adapt to new tasks.
As shown in the figure below, this is an example of image classification based on meta-learning. Suppose the target task is to train a model to classify horses, but there is relatively little labeled data for horses. Therefore, we can first find some auxiliary tasks with a large amount of data, such as cat classification, dog classification, etc., and use meta-learning algorithms to train on these auxiliary tasks, learn how to learn new tasks, and obtain a meta-learner. Then use this meta-learner as the initial model, and train it for several steps with a small amount of labeled horse data to quickly obtain a horse classifier. Obviously, the premise for meta-learning to work is that the auxiliary tasks used must be close enough to the target tasks.
How to apply meta-learning to the scenario of Fitness prediction?First of all, our goal is to rank the mutations of the target protein by Fitness, and the model to be trained is PLM using LoRA technology.
We adopted two strategies to construct auxiliary tasks. The first one is to find mutation experiment datasets of similar proteins in the existing DMS database based on the similarity with the target protein, and select the first two datasets as two auxiliary tasks.The starting point for doing this is to consider that the Fitness Landscape of similar proteins is also similar.
The second strategy is to use the MSA model to score candidate mutations of the target protein to form a pseudo-label dataset and use it as the third auxiliary task.The reason why we choose the MSA model is that the mutation prediction effect of the MSA model is usually not inferior to that of the PLM. We hope to perform data enhancement through MSA and give full play to the representation ability of the PLM.
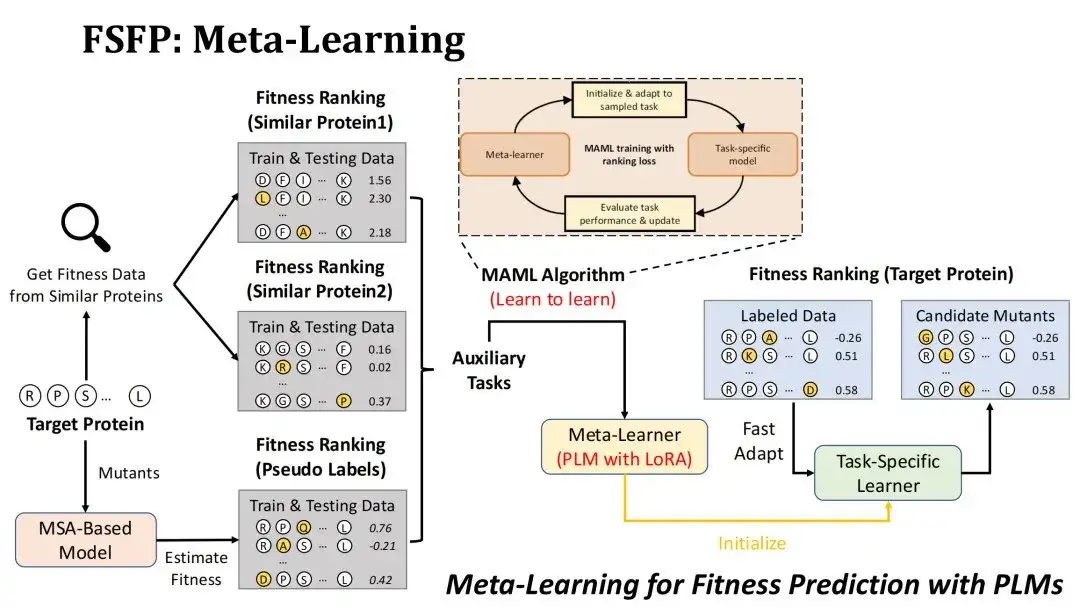
The meta-learning algorithm we use is MAML, whose training goal is to make the test loss of the meta-learner as small as possible after fine-tuning k steps with the training data of an auxiliary task, so that it can roughly converge after fine-tuning k steps on the target task.
Performance evaluation of FSFP method in protein fitness prediction
Benchmark creation
Our Benchmark data comes from ProteinGym, which originally contained 87 DMS datasets and has now been updated to 217.The proteins corresponding to 87 DMSs are roughly divided into four categories: eukaryotes, prokaryotes, humans, and viruses, covering a total of about 15 million mutations and corresponding Fitness.
For each dataset, we randomly selected 20, 40, 60, 80, and 100 single-point mutations as small sample training sets, and the remaining mutations as test sets. It should be noted that we did not use an additional validation set for early stopping, but instead estimated the number of training steps through cross-validation on the training set.

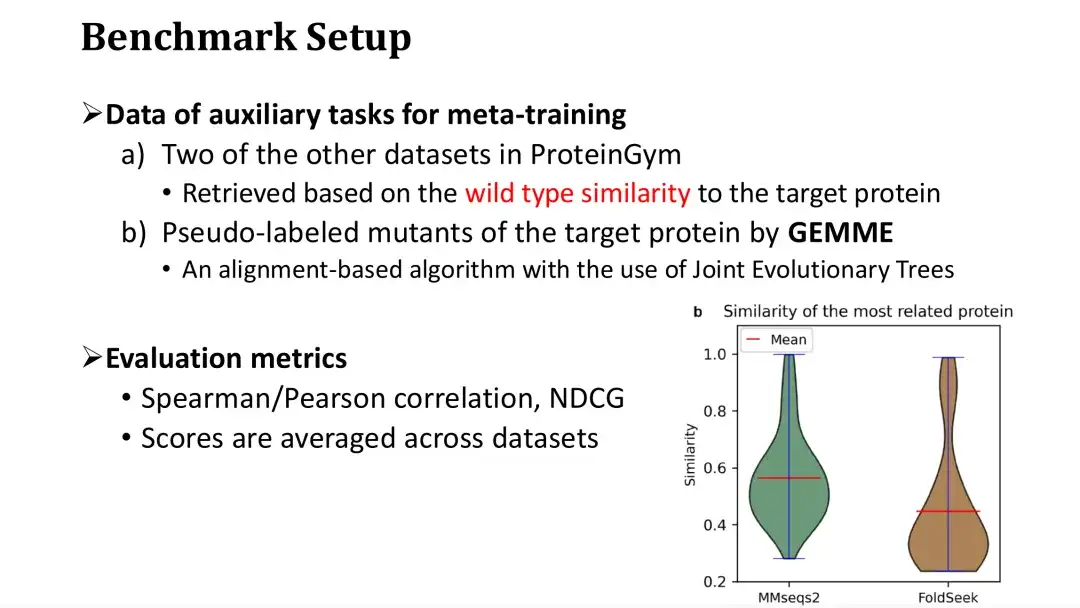
It was mentioned earlier that meta-learning requires three auxiliary tasks, two of which are retrieved from the DMS database based on their similarity to the target protein.When training on a dataset, we retrieve from the rest of the datasets in ProteinGym, assuming it is the database.
As shown in the figure below, each protein in ProteinGym is used as a query, and the similarity distribution of the most similar proteins is retrieved by MMseqs2 and FoldSeek. It can be seen that the average sequence or structural similarity of the most similar proteins is around 0.5. The third auxiliary task involves scoring mutations using the MSA model. We chose the GEMME model, which constructs an evolutionary tree based on MSA and calculates the conservation of each point on the evolutionary tree to score mutations.
The evaluation indicators used are Spearman/Pearson coefficient and NDCG, which are common evaluation criteria in Fitness prediction tasks. The final evaluation score is the average score on 87 data sets.
Ablation experiment of FSFP on ESM-2
As shown in the figure below, the x-axis in the left figure represents the size of the training set, and the y-axis represents the Spearman coefficient. Each line corresponds to a different model configuration. The top line represents the complete FSFP model; the second line represents the replacement of the third auxiliary task of meta-learning with DMS data of similar proteins without using MSA. It can be seen that the model performance has decreased after removing the MSA information; the third line indicates that the Spearman coefficient has further decreased when meta-learning is not used and only ranking learning and LoRA are relied on.
The green line represents the ridge regression model previously published in NBT, which is one of the few baseline models currently suitable for small sample scenarios; the gray dotted line represents the zero-shot score of ESM-2; the bottom two lines represent the results of training ESM-2 using traditional regression methods.
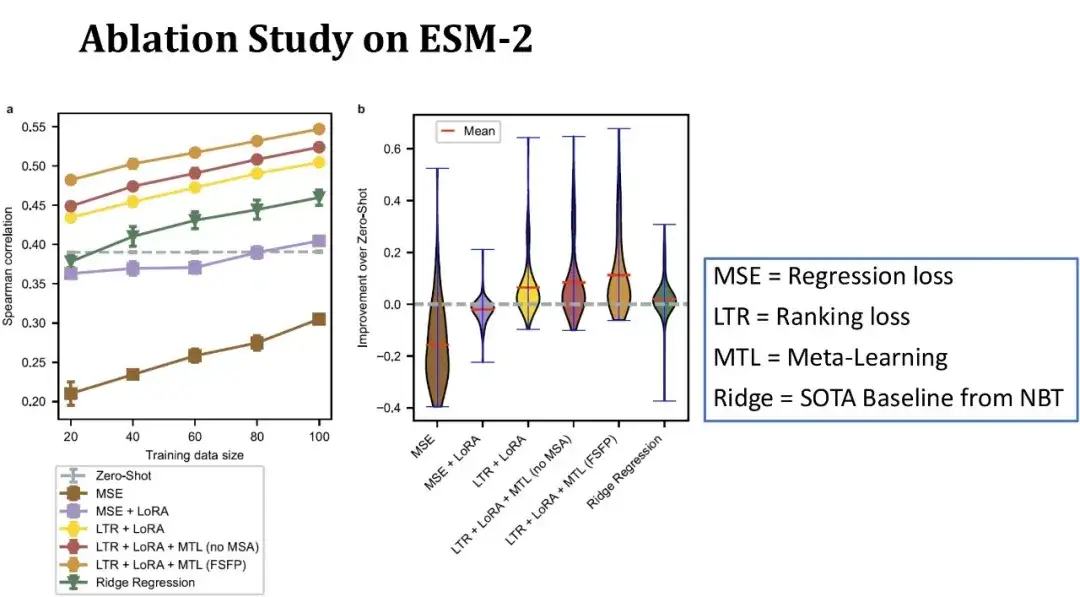
Overall, with only 20 training samples, our method improves the Spearman by 10 points compared to zero-shot, and each module plays a positive role in model performance. The figure on the right shows the distribution of performance improvement compared to zero-shot on 87 datasets, with a training set size of 40 samples.It can be seen that our method can improve model performance on most datasets, and the improvement on some datasets even exceeds 40 points, showing more stable performance than the baseline.
Effectiveness of Meta-Learning
The purpose of meta-learning is to enable PLM to converge quickly on the target task with a small number of iterations.The following are some examples to illustrate this.
The following three charts show the training curves of fine-tuning on three datasets using 40 training samples. The x-axis represents the number of training steps, and the y-axis represents the Spearman coefficient on the test set. The top orange and red lines are models trained with meta-learning, the former uses MSA to build auxiliary tasks, and the latter does not. The yellow line represents a model that only uses ranking learning and LoRA without meta-learning.
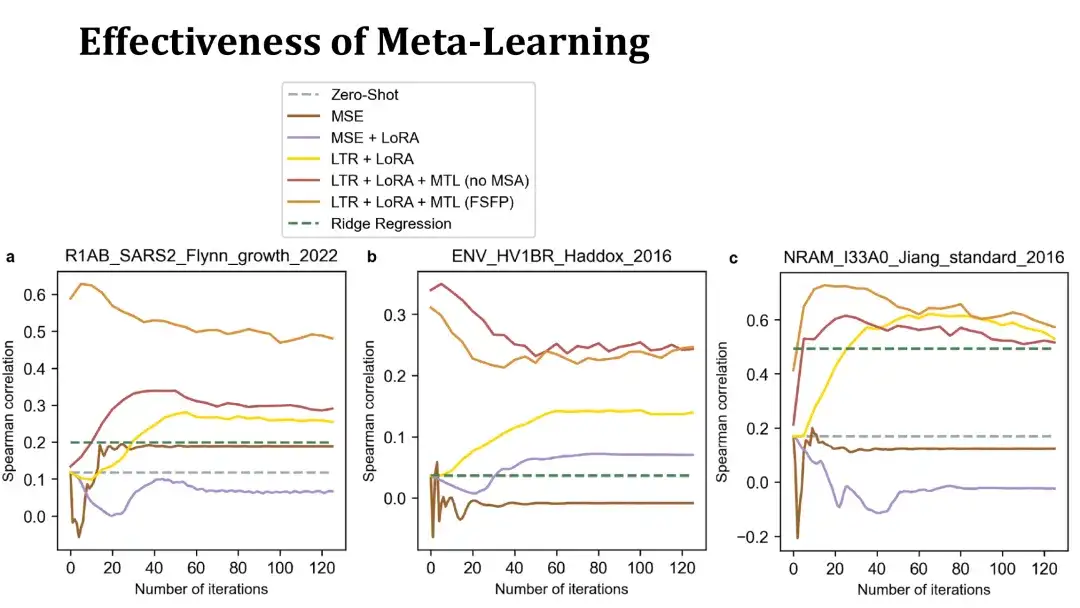
As you can see,The meta-learning trained model can improve performance more quickly on the target protein and reach a high score within 20 steps, sometimes even better than the initial model without fine-tuning. This shows that meta-learning has obtained an effective initial model.The MSE-based model below performs poorly and overfits quickly, making it difficult to surpass the zero-shot method.
Results of applying FSFP to different PLMs
We selected three typical PLMs, namely ESM-1v, ESM-2 and SaProt.The first two models only use protein sequence information, while SaProt combines protein tertiary structure tokens.
The line graph on the left shows the Spearman score for predicting the effect of a single point mutation under different training set sizes. The same color represents the same model, and different shapes of points represent different training methods. The dots above represent the FSFP method, the inverted triangle below represents the ridge regression, and the dotted line represents the zero-shot performance of the model. The purple line represents the GEMME model, which is not a PLM, but the ridge regression method can be combined with it.It can be seen that the FSFP method can steadily improve the performance of each PLM, and is much better than the ridge regression and the zero-shot of the corresponding model.
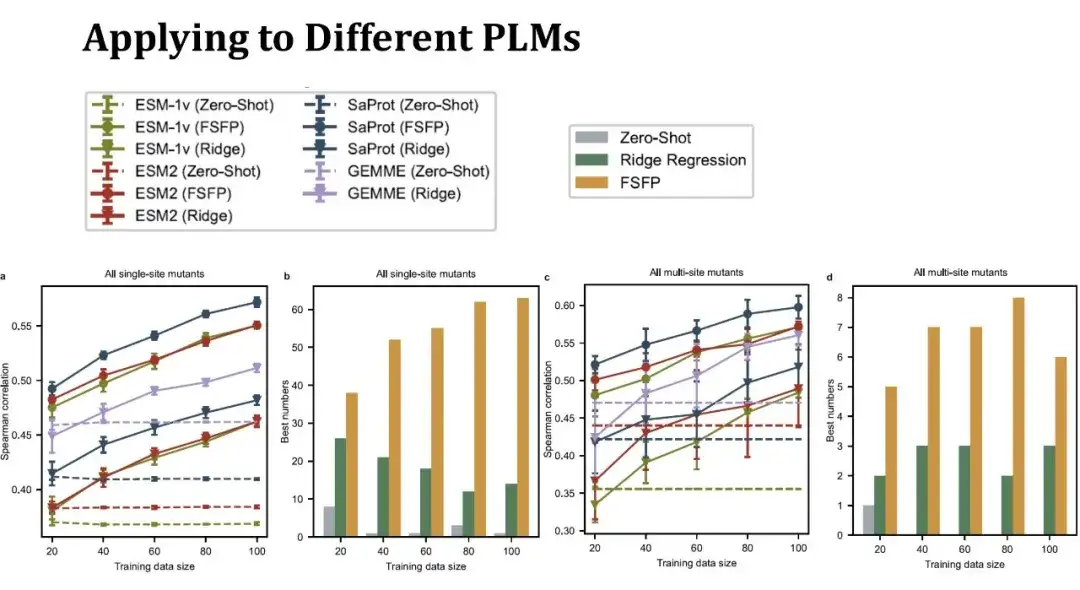
The second bar graph shows the number of highest scores obtained using the three strategies (zero-shot, ridge regression, and FSFP) on different datasets. FSFP performs best in most datasets.The two figures on the right show the performance of predicting multi-point mutations. There are 11 multi-point mutation data sets involved, and the conclusions are similar to those of single-point mutations. However, the variance of the ridge regression model is larger here, indicating that it is more sensitive to data segmentation.
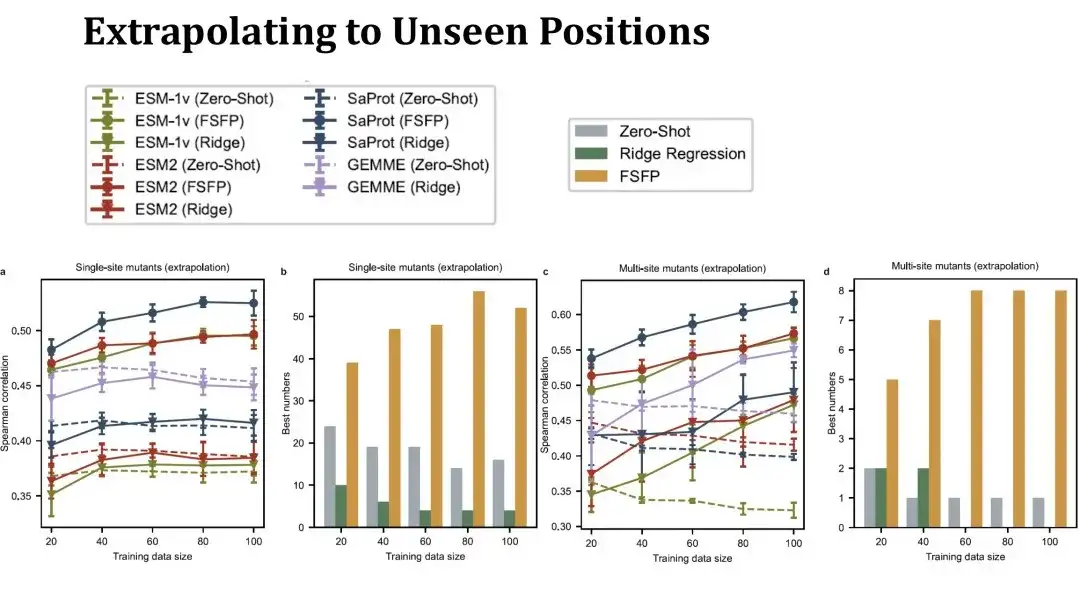
We then evaluated the extrapolation performance of FSFP, specifically evaluating the prediction performance at mutation sites not seen in the training set.In this case, the test set will be much smaller than before, and the test set will change significantly as the training set gets larger, so the zero-shot performance in the table is no longer a straight line. This setting is more challenging. You can see that the performance of single-point mutation ridge regression on the left can hardly exceed zero-shot, but FSFP can still steadily improve the performance. The test results of multi-point mutation on the right also show that our training method has good generalization ability.
Transforming Phi29 with FSFP
In addition, we also used FSFP to perform a case study on protein modification.The target protein is Phi29, a DNA polymerase, and we hope to improve its Tm by single-point mutation.
The experimental process is as follows: first, use ESM-1v to perform zero-shot scoring on saturated single-point mutations, select the top 20 mutations and perform wet experiments to measure Tm; then use these 20 experimental data as a training set, use FSFP to train ESM-1v, use the trained model to score the saturated single-point mutations again, and reselect the top 20 mutations for testing.
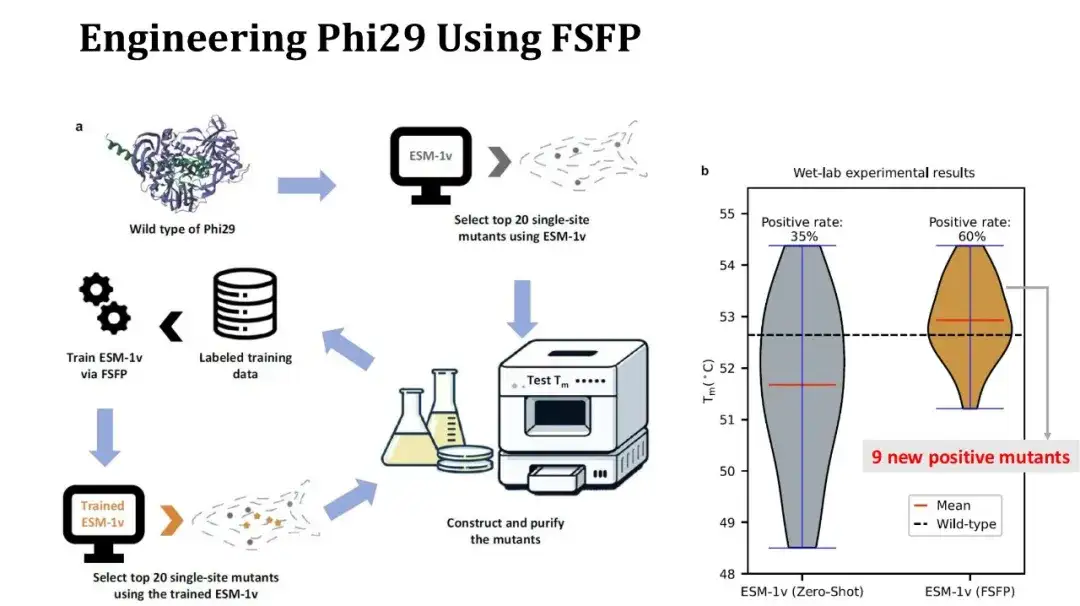
The right figure shows the Tm distribution comparison of the first and second rounds of experiments. 7 out of 20 mutations in the first round were positive, which increased to 12 in the second round, and the average Tm increased by 1 degree. Among them, 9 of the positive mutations found in the second round were new. Although the positive rate and average Tm have increased, unfortunately the highest Tm has not increased, because the mutation with the highest Tm obtained in the second round still exists in the first round results. However, since more positive single-point mutations have been obtained, we can try to combine these sites for high-point mutation experiments to further improve Tm.
Summary of FSFP Method and Future Research Prospects
FSFP is a small sample learning strategy for PLM, which can significantly improve the performance of PLM in mutation effect prediction using a small number (dozens) of labeled training samples, and can be flexibly applied to a variety of different PLMs.Experiments show that the design of FSFP is reasonable:
* Ranking learning meets the basic requirement of mutation ranking in protein engineering and reduces the difficulty of training;
* LoRA reduces the risk of overfitting by controlling the amount of trainable parameters of PLM;
* Meta-learning can provide good initial parameters for the model, enabling the model to quickly migrate to the target task.

Finally, we discuss the future directions of AI-assisted directed evolution. The general process of AI-assisted directed evolution is to start with a set of initial mutations, obtain their Fitness Labels through wet experiments, and use the labeled data fed back from the experiments to train the machine learning model. Then, the next round of mutations to be tested are selected based on the model's predictions, and the process is repeated.
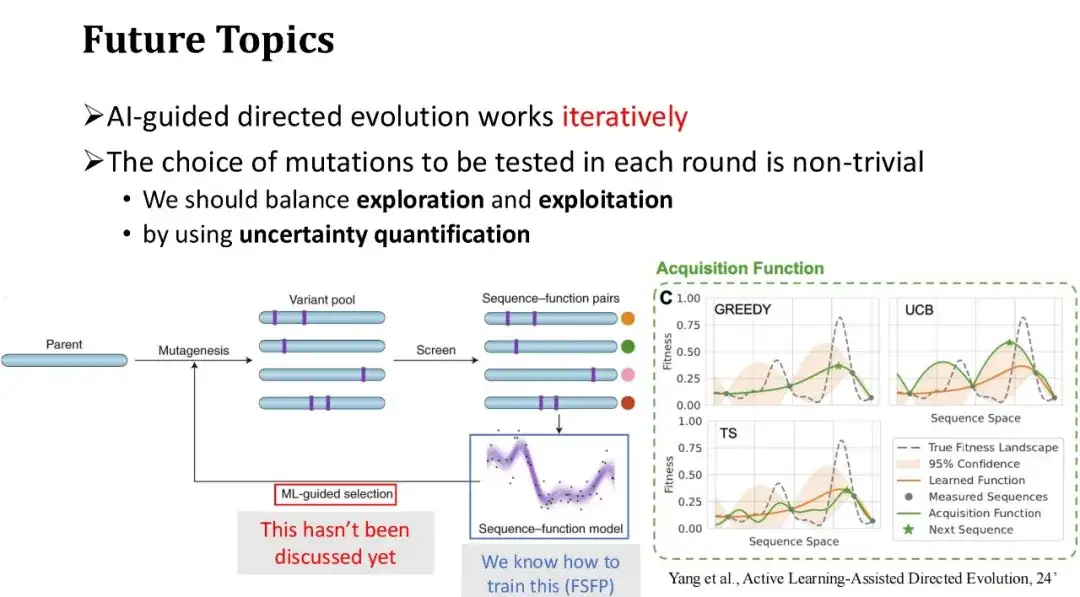
FSFP mainly solves the problem of small sample training of the model in each round of experimental iteration and improves the prediction accuracy of the model.However, we have not yet discussed how to effectively select mutations to be tested in the next round, that is, the new training samples to be added in the next round. In the previous example of Phi29 protein modification, we directly selected the top 20 mutations with the highest model scores. However, in the scenario of multiple rounds of iterations, the greedy selection strategy is not necessarily the best method, and it is easy to fall into the local optimum. Therefore, it is necessary to find a balance between exploration and utilization.
In fact, the process of iteratively selecting test samples to label and gradually expanding the training data is an active learning problem, which has made some research progress in the field of protein engineering. For example, Frances H. Arnold, an authoritative scientist in the field of directed evolution, discussed related issues in her article "Active Learning-Assisted Directed Evolution".
Paper address:
https://www.biorxiv.org/content/10.1101/2024.07.27.605457v1.full.pdf
We can use uncertainty quantification techniques to assess the uncertainty of the model's scoring of each mutant. Based on these uncertainties, the selection strategy of test samples will be more diverse.A commonly used strategy is the UCB method, which selects the mutation samples with the highest model prediction uncertainty for the next round of annotation, that is, it gives priority to samples with the largest prediction variance. This is similar to the human learning process: if we do not have a good grasp of certain knowledge points or are uncertain, we will focus on strengthening our learning.








