Command Palette
Search for a command to run...
Sensitivity Improved by 56%, CUHK/Fudan/Yale and Others Jointly Proposed a New Protein Homolog Detection Method

Protein is the material basis of life and the main bearer of life activities. In the post-genomic era, with the development of protein determination technology, the size of protein sequence database has exploded. In order to gain a deeper understanding of the diversity and function of proteins, protein identification is particularly important in biology.
In the process of protein recognition, protein sequence homology identification is one of the most important tasks.It can help scientists understand the evolutionary relationships, structural features, and functions of proteins. Although traditional protein sequence alignment methods perform well in many cases, they are powerless when faced with distant homologs. These distant homologs are often ignored in conventional alignments due to their low sequence similarity, thus limiting researchers' comprehensive understanding of protein diversity and complexity.
To solve the pain points of distant protein homology research, based on protein language models and dense retrieval technology, Li Yu from the Chinese University of Hong Kong, together with Sun Siqi, a young researcher from the Laboratory of Intelligent Complex Systems at Fudan University and the Shanghai Artificial Intelligence Laboratory, and Mark Gerstein from Yale University, proposed an ultra-fast and highly sensitive homology detection framework - the Dense Homology Retriever (DHR).
DHR can identify distant homologs hidden deep in the sequence without relying on traditional sequence alignment, through the powerful capabilities of the dual encoder structure and protein language model, bringing unprecedented speed and sensitivity to homolog identification. The study was published in the internationally renowned journal Nature Biotechnology under the title "Fast, sensitive detection of protein homologs using deep dense retrieval".
Research highlights:
* Compared with previous methods, DHR improves sensitivity by more than 10% and improves sensitivity by more than 56% at the superfamily level for samples that are difficult to identify using alignment-based methods
* DHR code queries sequences and databases 22 times faster than traditional methods such as PSI-BLAST and DIAMOND, and 28,700 times faster than HMMER

Paper address:
https://doi.org/10.1038/s41587-024-02353-6
The open source project "awesome-ai4s" brings together more than 100 AI4S paper interpretations and provides massive data sets and tools:
https://github.com/hyperai/awesome-ai4s
Constructing datasets in multiple dimensions to explore a wider spectrum of protein sequences
The training set constructed in this study includes 2 million query sequences carefully selected from UR90.Using the JackHMMER algorithm, the study iteratively searched for candidate sequences in Uni-Clust30 and aligned the candidate sequences with multiple sequence alignments (MSAs). Each MSA contained 1,000 homologs to ensure that only the most relevant sequences were retained. After rigorous screening, JackHMMER was redeployed to process the different sequences obtained and used the same hyperparameter settings as AF2 (AlphaFold 2) to facilitate fair comparison.
In the study of large data sets, the study selected the BFD/MGnify data set.This is a massive database of approximately 300 million proteins, enabling exploration of a wider spectrum of protein sequences.
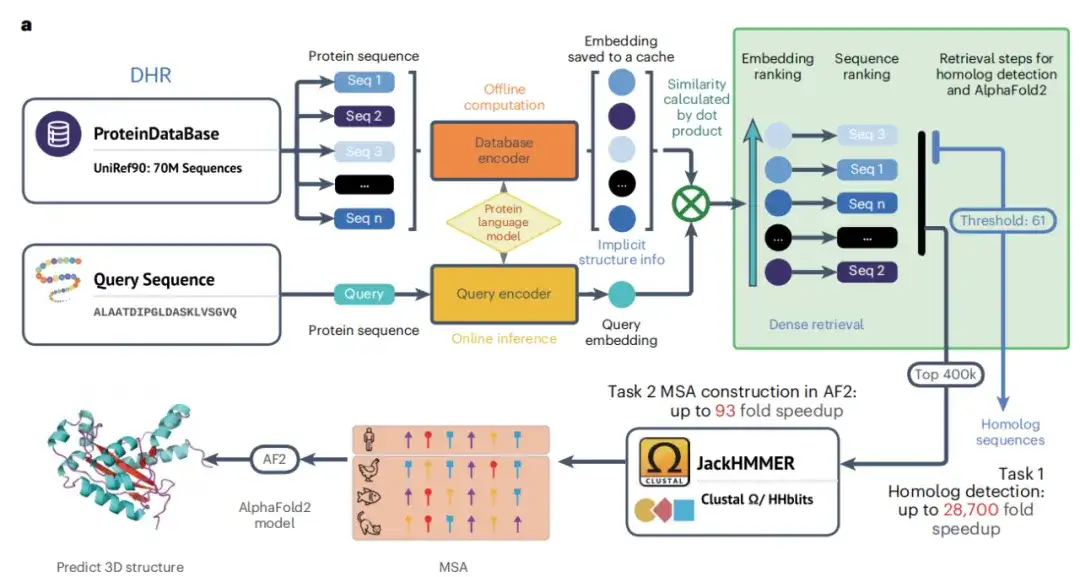
The DHR method: an ultrafast and sensitive protein homology search pipeline
The core idea of the DHR method is to encode protein sequences into dense embedding vectors so as to effectively calculate the similarity between sequences.Specifically, this study effectively trained the sequence encoder by initializing ESM and integrating contrastive learning techniques, thereby creating conditions for the construction of a protein language model and enabling DHR to be used more effectively to retrieve homologs.
As shown in Figure a below, with the completion of the dual encoder training phase, the study was able to generate high-quality offline protein sequence embeddings. The study then used these embeddings and similarity search algorithms to retrieve homologs for each query protein. By specifying similarity as a retrieval metric, similar proteins can be found more accurately than traditional methods, and the similarity between the two proteins is used for further analysis. Finally, JackHMMER constructs the MSA of the retrieved homologs, and the study obtained the DHR technology that can quickly and effectively discover homologs.
Not only that, the study also developed a hybrid model DHR-meta, which outperformed the individual pipelines on CASP13DM (domain sequence) and CASP14DM targets by combining DHR and AF2 default.
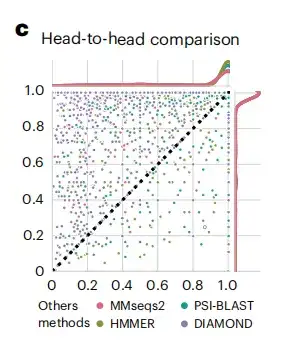
After obtaining the generated protein embeddings, the study evaluated the performance of DHR by comparing it with methods on the standard SCOPe (Structure Classification of Proteins) dataset.As shown in Figure c below, the sensitivity of DHR data is better than other methods.
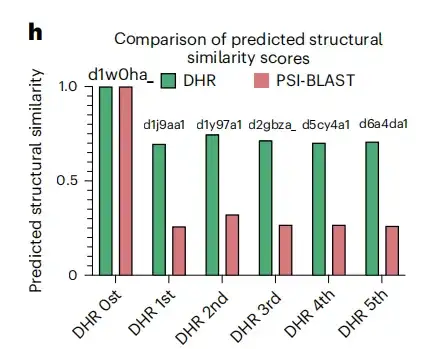
In addition, as shown in Figure h below, in the specific example of the d1w0ha query, neither PSI-BLAST nor MMseqs2 matched any results, but DHR retrieved 5 homologs, which were also classified as the same family as d1w0ha in SCOPe. This means that DHR can capture more structural information. Compared with traditional methods such as PSI-BLAST, MMseqs2, DIAMOND, and HMMER, DHR detected the most homologs (with a sensitivity of 93%).This demonstrates that DHR is able to integrate rich structural information and achieve 100% sensitivity in many cases.
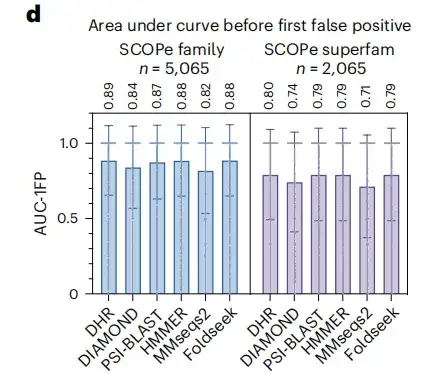
To strengthen the credibility of the research results, the study also included another standard indicator, the area under the curve before the first FP. The results showed that, as shown in Figure d below, DHR reached a score of 89%.Meanwhile, other methods also showed comparable performance to DHR, but their execution time was significantly longer.When the study further analyzed the superfamily level of more challenging distant homologs, all methods experienced a significant performance drop, with an overall drop of approximately 10%. Despite this, DHR still maintained its leading performance with an AUC-1FP score of 80%.
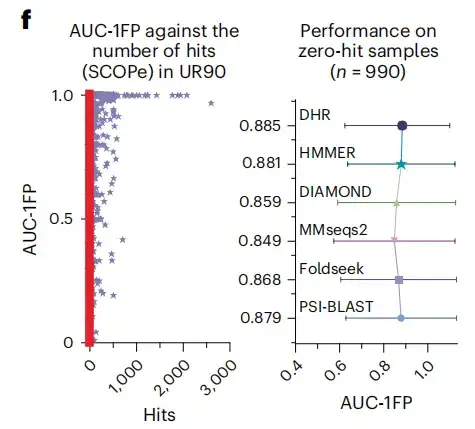
The study also found that when using BLAST to compare the SCOPe database and UniRef90, most samples produced less than 100 matches, and even about 500 samples did not get any matches, indicating that these samples were "unseen" structures in the training dataset. In contrast, DHR still achieved high-quality predictions when facing these structures, reaching an AUC-1FP score of 89%.This demonstrates that DHR is capable of handling completely new data.
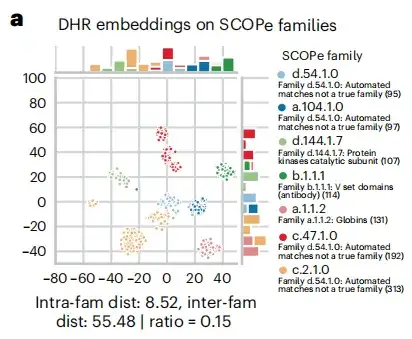
In the homology search process, as shown in Figure a below, the study found that DHR sequence embedding contains a lot of structural information, and the accuracy of DHR in retrieving homologs even exceeds that of structure-based alignment methods. Based on this result,This study further revealed the correlation between sequence similarity ranking and structural similarity of DHR.
Research results: DHR has better accuracy and effectiveness, and can build high-quality MSA on large-scale datasets
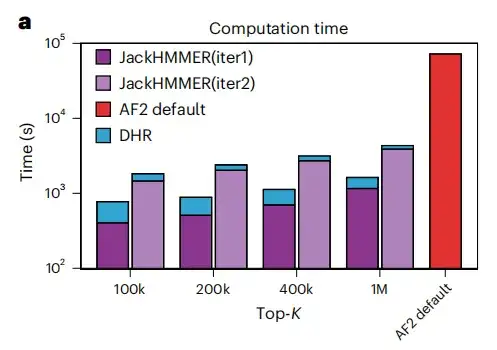
This study used homologs provided by DHR to create MSA from JackHMMER and compared it with the AF2 default pipeline. As shown in Figure a below, the average running speed of all configurations of DHR + JackHMMER is faster than the ordinary JackHMMER of AF2. Moreover, DHR overlaps with JackHMMER by about 80% when building MSA on UniRef90,This suggests that many downstream tasks related to MSA can be performed using DHR, producing similar results but faster.
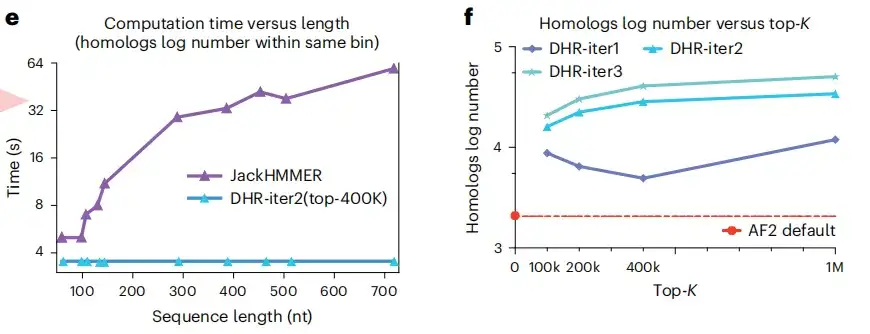
As shown in Figures e and f below, another advantage of DHR is that it can construct the same number of homologs of different lengths in a constant time, while JackHMMER is linearly scalable. Moreover, compared with AF2, DHR can also provide more homologs and MSA for query embedding. These results all show thatDHR is a promising approach for all categories of MSA construction.
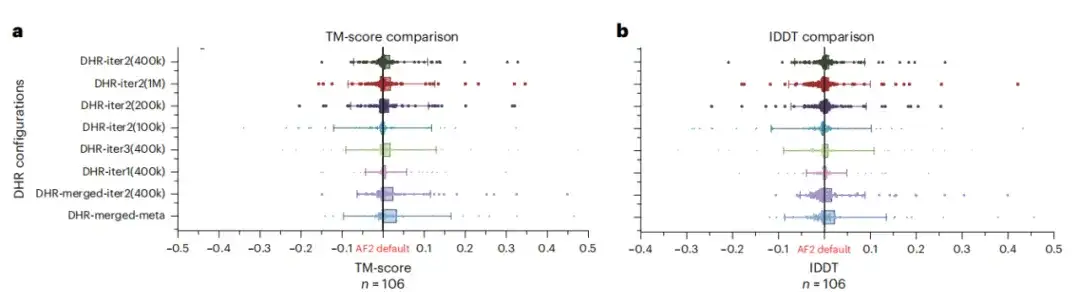
Although DHR can generate different MSAs, this study further analyzed whether it can be used as an MSA supplement to the AF2 baseline. The results show that merging all MSAs with AF2 under different DHR settings has the best performance, as shown in Figures a and b below.This means that DHR can quickly and accurately replenish AF2's MSA pipeline.
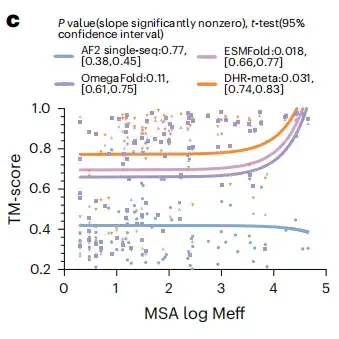
To test the potential benefits of large language models for protein structure prediction, the study evaluated whether replacing MSA with a large language model on all CASP14DM targets would produce better results. As shown in Figure c below, in simple cases with a large number of available MSAs, the language model can convey as much information as MSA. But as the sequence length increases, DHR-meta performs better and better, outperforming ESMFold in almost all cases. This means that compared with language model-based methods,The MSA-based model can greatly improve the accuracy and effectiveness of predictions.
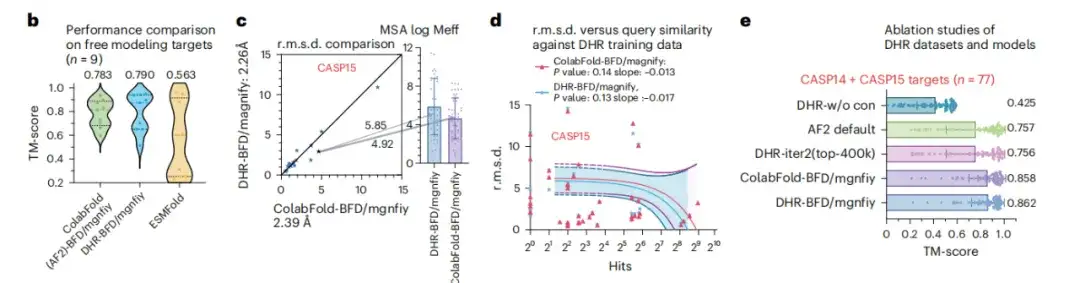
In order to study the scalability of DHR in large data sets, this study conducted an in-depth analysis of DHR based on BFM/MGnify. As shown in Figure b below, in the complex scenario of predicting the structure of FM targets, DHR was able to stand out by generating more meaningful MSAs, and the performance of the ColabFold method using MMseqs2 to construct MSAs was 0.007 TM-score higher.
In Figure c, DHR shows a slight performance improvement relative to ColabFold-MMseqs2. Figure d also shows that after similarity testing on CASP14 and SCOPe, it was found that DHR did not simply remember the query or hit results, but performed a comprehensive similarity assessment on all targets. These results all prove thatDHR enables the construction of MSAs of disordered proteins on large-scale search datasets with high diversity.
Young forces in the field of protein structure prediction
Undoubtedly, protein structure prediction plays an important role in drug development, antibody design and other applications. AI may become the key to solving the historical problem of limited accuracy of protein structure prediction. In this key field, domestic scientific research teams have gradually formed a trend of a hundred schools of thought, and rising young researchers have become a force that cannot be ignored. Li Yu and Sun Siqi, who led the above research results, are both outstanding.

Li Yu received his bachelor's degree (honors) in biological sciences from the Bei Shizhang Elite Class of the University of Science and Technology of China in 2015, his master's degree in computer science from King Abdullah University of Science and Technology (KAUST) in Saudi Arabia in December 2016, and his doctorate in computer science from the same university in 2020.
In December of the same year, he returned to China and joined the Department of Computer Science and Engineering of the Chinese University of Hong Kong as an assistant professor, leading the Artificial Intelligence in Healthcare (AIH) group. He conducted in-depth research at the intersection of machine learning, healthcare, and bioinformatics, and led the team to develop new machine learning methods to solve computational problems in biology and healthcare, especially structured learning problems.
Regarding the fields of biology and healthcare in which he has been deeply involved, Li Yu said, "My long-term goal is to improve the healthcare system and directly benefit society by improving people's health and well-being."It is worth mentioning that he was also selected into the 2022 Forbes Asia "30 Under 30" list (Healthcare and Science).

Sun Siqi has won excellent results in the global protein structure prediction competition and is currently a young researcher at the Basic Theory and Key Technology Laboratory of Intelligent Complex Systems and the Shanghai Artificial Intelligence Laboratory at Fudan University.He is committed to the application research of deep learning in interdisciplinary fields such as life sciences and natural language processing, and focuses on improving the accuracy and speed of models and solving specific problems in the implementation of models.
In terms of protein prediction, he focuses on predicting the structure and sequence of proteins through deep learning models, and training models to identify patterns and regularities in sequences to predict the sequence and folding of proteins, thereby improving the accuracy and efficiency of protein de novo sequencing and structure prediction, and creating new possibilities for drug design and disease treatment.
In the field of AI4S in China, more and more young people are active. It is foreseeable that AI technology will play a more critical role in the field of protein structure prediction, but the road ahead is long and arduous. Fortunately, domestic scientific research teams have demonstrated an indomitable spirit of exploration and innovation, not only working hard on algorithm optimization and model construction, but also conducting in-depth research in data processing and experimental verification to ensure the scientificity and practicality of research results. These efforts are gradually being transformed into practical applications, bringing new vitality and hope to the fields of pharmaceutical research and development, biotechnology, etc.
Finally, I recommend an academic sharing activity!
The third live broadcast of Meet AI4S invited Zhou Ziyi, a postdoctoral fellow at the Institute of Natural Sciences of Shanghai Jiao Tong University and Shanghai National Center for Applied Mathematics. Click here to make an appointment to watch the live broadcast!
https://hdxu.cn/6Bjomhdxu.cn/6Bjom