Command Palette
Search for a command to run...
Dr. Bingxin Zhou From Shanghai Jiao Tong University: Anchoring the Challenge of Scarce Biological Data, Graph Neural Networks Reshape Protein Understanding and Generation

On August 12, the Shanghai Jiao Tong University AI for Bioengineering Summer School officially opened, attracting more than 100 industry professionals from more than 30 universities and 27 companies at home and abroad. During the three-day learning and exchange, many industry experts, business representatives and outstanding young scholars,In-depth sharing was provided on the integration and innovative development of AI and bioengineering.
On the morning of the 12th, Zhou Bingxin, assistant researcher at the Institute of Natural Sciences of Shanghai Jiao Tong University and Shanghai National Center for Applied Mathematics (Shanghai Jiao Tong University Branch), spoke on the theme of "The Past and Present of Artificial Intelligence". It vividly sorts out the development history of AI and summarizes the characteristics of the milestone model.
In the afternoon, in the guest expert report of "Frontier Progress of Artificial Intelligence", Dr. Zhou Bingxin also spoke on the theme of "Graph Neural Networks and Protein Structure Representation". He shared with everyone the definition, advantages, and cutting-edge applications of graph neural networks in areas such as protein prediction and generation. HyperAI has compiled and summarized Dr. Zhou Bingxin's speech without violating the original intention. The following is the transcript of the speech.
After decades of rapid development, deep learning has produced various models such as convolutional neural networks, recursive neural networks, and Transformers, which can be used to process data with different characteristics. Among them, graph neural networks are widely used in social networks, trajectory prediction, molecular modeling and other scenarios because they can input and process structural data.
However, many people think that graph neural networks are graph convolutional networks (GCNs), which cannot fit complex functions and have many limitations, such as oversmoothing problems when multiple layers are stacked. In addition, since large models based on Transformer have strong learning capabilities on large data sets,So why do we continue to research and develop graph neural networks?
To these questions, I summarize the answer as: "It's SEXY".
The first "S" is that research based on graph neural networks is healthy and sustainable. As shown in the figure below, by comparing the carbon consumption of various human behaviors, we can see that the premise of large models having powerful capabilities is huge energy consumption. In addition, excessive concentration of computing resources and research focus on large models will also squeeze out the living space of other model research. In the long run, only large companies that monopolize computing resources or discourse power can maintain artificial intelligence research and development, and the research space of researchers from non-large companies will be greatly limited.
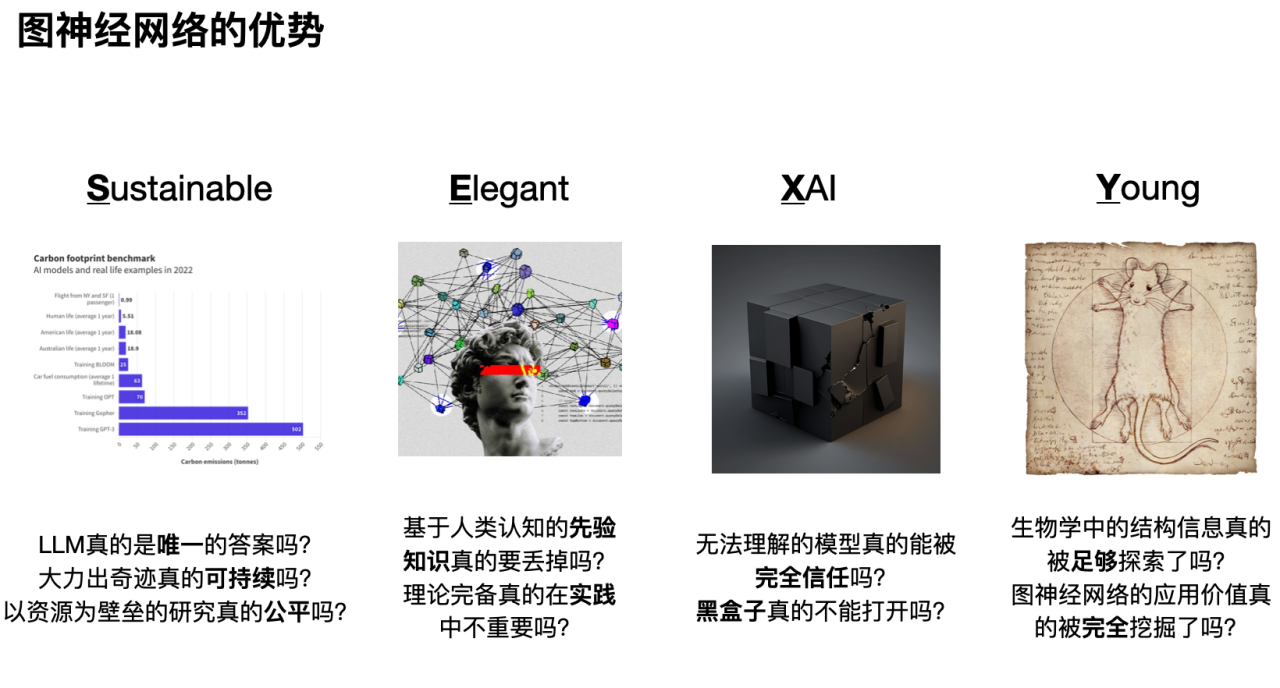
The second "E" is that hundreds of years of accumulation in natural science should not be ignored by the rapid development of artificial intelligence. In addition to learning feature representation, graph neural networks can also elegantly incorporate prior knowledge from humans (inductive bias). In addition, compared with other data-driven models, graph neural networks have more theoretical support, such as signal processing, social dynamics, etc.
The third “X”, graph neural networks help promote the interpretability of deep learning networks. With the development of artificial intelligence, people are paying more and more attention to the significance and rationality of model results. Through in-depth research on the interpretability of graph neural networks, we can better understand the logic and basis behind model decisions and improve the reliability and trust of the model.
Fourth, “Y”: As a young and rapidly developing field, graph neural networks still have a large number of unresolved problems and challenges, providing researchers with a broad space for exploration. In addition, just like convolutional neural networks for image processing and self-attention mechanisms for natural language processing, graph neural networks will also provide good solutions to many biological problems (especially those with insufficient data and important prior knowledge).
Next, I will share with you the specific application value of graph neural networks from three aspects: molecular data and graph representation, introduction to classic graph neural networks, and graph neural networks and more biological problems.
Molecular Data and Graph Representation: Three Elements of Biological Data Graphics
To convert biological data into a graph representation, we must first answer: what is a graph and what basic elements does it consist of. Generally speaking,A graph contains three elements: nodes; edges (connection relationships between nodes); graphs (the complete entity composed of nodes and edges).
How do we use these three elements to define the object of study in biology? The following figure shows four examples:

For a small molecule (Fig. Each atom can be defined as a node, and the distance relationship or chemical bond relationship between atoms can be represented by edges.
If we look at protein at the amino acid level, The protein as a whole can be viewed as a graph, with each amino acid as a node in the graph. When different amino acids are close in spatial position, it can be assumed that there is some correlation between them, and these spatially close amino acid nodes are connected through edges.
Similarly, if we look at proteins based on their secondary structure, Then each secondary structure can be regarded as a node in the protein graph, and its adjacent or spatially close secondary structures are connected by edges.
Finally, for the disease knowledge graph, Different diseases, genes, medications, patients and other elements can be regarded as nodes, and the connections between nodes represent the complex relationships between them, such as a certain drug can treat a specific disease, or a certain gene causes a certain disease.
After defining a graph,The next step is to consider how to describe the information on the graph, such as the characteristics of nodes and edges?
As shown in the figure below, there are certain relationships between the four nodes. In order to accurately describe these relationships, an adjacency matrix A can be defined. When processing different biological data, the adjacency matrix can be used to characterize whether there are covalent bonds between atoms or to determine the k-order neighbors of a certain amino acid.

Additionally, each node and edge can have a set of attributes attached to it. Taking amino acid nodes as an example, node attributes may include characteristic information such as its type, physical and chemical properties, etc. Edges, as bridges connecting nodes, can also carry characteristic information, such as the characteristic vector on each edge, which covers the distance between two amino acids (including sequence distance and spatial distance), the basis for establishing the edge (based on spatial structure or atomic chemical bonds, etc.). These edge features provide a more detailed and in-depth perspective for understanding the relationship between nodes.
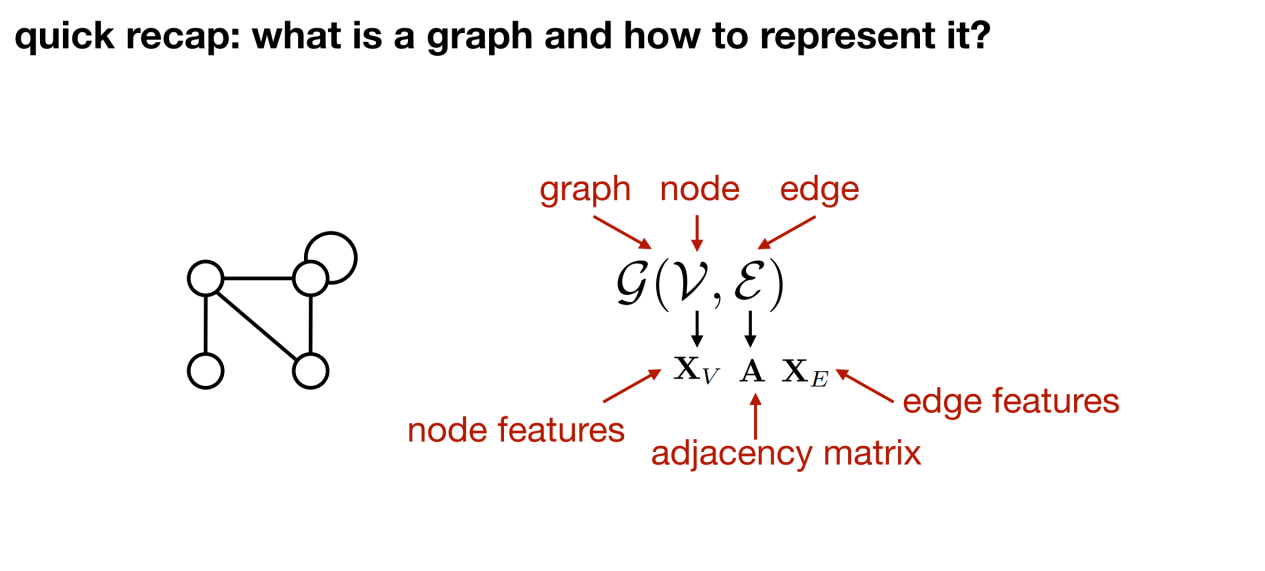
In summary,Every structured entity (such as a protein) can be represented as a graph. As shown in the figure below: G can be used to represent the graph, v represents the node, ε represents the edge, Xv represents the features on the node, the adjacency matrix A represents the node connection, and Xe represents the features of the edge.
Based on the three basic elements of a graph (nodes, edges, and graphs), the vector representation and prediction tasks on the graph can be classified as follows:
- Node-level prediction. For example, when doing protein sequence design, given a known protein graph, predict the type of amino acid represented by each node in the graph.
- Link prediction. Given a graph and all nodes, infer whether there is a relationship between nodes, such as gene regulatory networks, drug knowledge graphs and other prediction tasks.
- Graph prediction (graph-level prediction). When both nodes and edges are determined, multiple graphs are learned and analyzed simultaneously to predict the labels of each graph.
What are graph neural networks: Not just GCN, but also GAT, GraphSAGE, EGNN, and more
Graph neural networks look for hidden representations of each node based on the connection relationships between given nodes, and find a vector representation for each node. Compared to other types of data, the biggest feature of graphs is that they can clearly indicate which nodes are directly related and the closeness of the relationships between different nodes.Therefore, the essence of graph neural networks lies in using these inductive biases and passing messages between connected nodes. The closer the neighbor nodes are, the greater their influence on the central node.
Next, I will share with you several classic graph convolutional neural networks.
The first one is the graph convolutional neural network GCN, As shown in the figure below, the core is that each layer of GCN will averagely aggregate the information of the first-order neighbors to the central node, and use the aggregated information as a new representation of the central node.
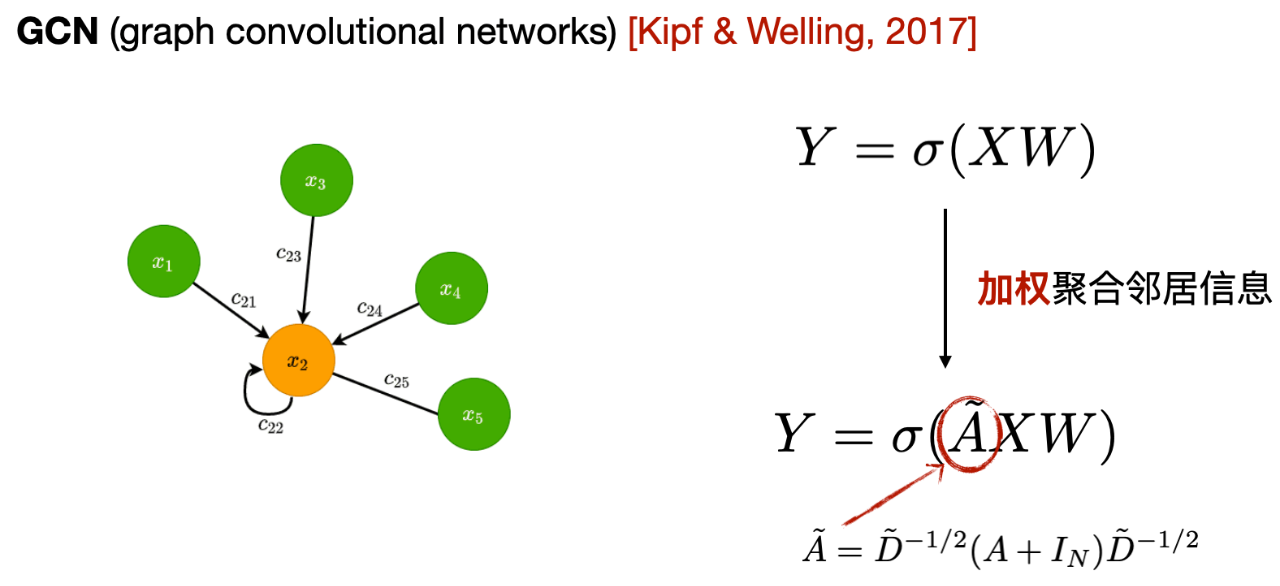
As can be seen from the expression, the difference between GCN and MLP is that GCN adds the adjacency matrix and uses the first-order neighbor information to update the node representation. In addition, it adds self-loops to strengthen its own information when aggregating information, and performs weighted average according to the number of neighbors of each neighbor node.
- First-order neighbors: The central node is directly connected to other nodes, that is, the points that can be reached through one edge are called first-order neighbors.
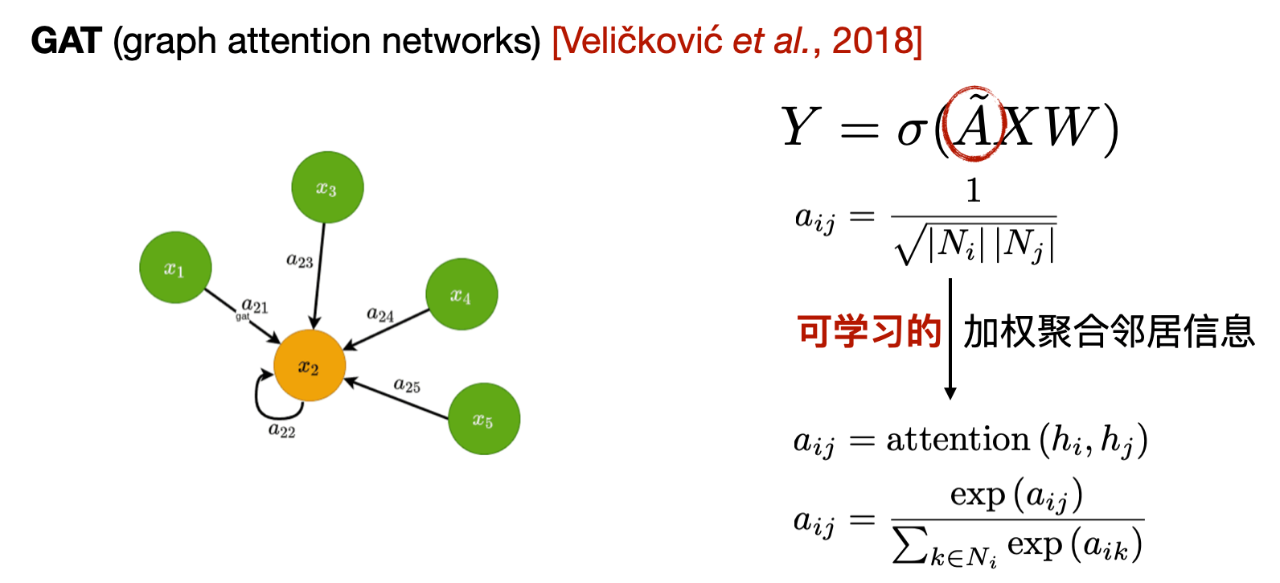
The second is the Graph Attention Network (GAT). Compared to GCN, the main difference of GAT is the way weights are calculated when aggregating neighbor information. GCN uses weights calculated based on the adjacency matrix, while GAT calculates a learnable weight based on the characteristics of neighbor nodes.
The above two methods are typical representatives of transductive methods. They require a complete graph as input, which increases the computational complexity.In this regard, GraphSAGE proposes an inductive approach. Each time information is transmitted, it is only necessary to understand the first-order neighbors of the central node, and only a part of the neighbor information is randomly selected for aggregation.
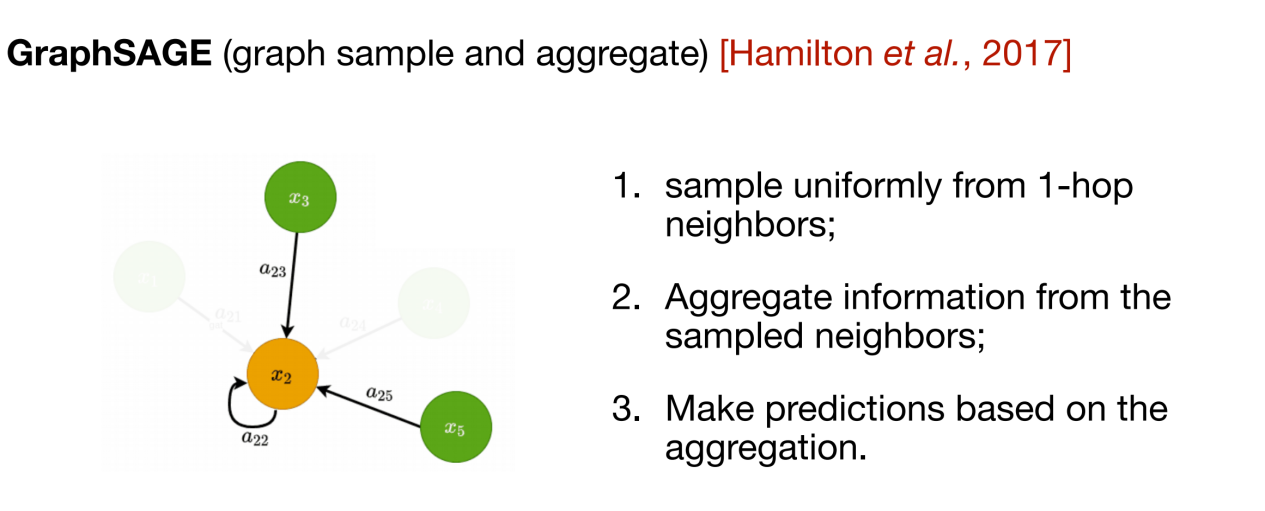
The above three methods are to update the representation of nodes in the two-dimensional topological structure graph, and the subsequent message passing network (MPNN) integrates this type of information aggregation method into a framework. However, many biological data (such as molecules) also need to consider the three-dimensional spatial structure.In order to integrate spatial information, equivariant graph neural network (EGNN) can be used. As shown in the figure below, the core of this method is that in addition to the feature information of the node itself, the relative position relationship between nodes is also introduced to ensure the rotational equivariance and translational invariance of the learned representation.

In addition, there are many advanced graph neural network designs. Some designs can not only improve the predictive performance of the model, but also focus on improving efficiency, reducing over-smoothing, adding multi-scale representation and other needs. By introducing continuous message passing, spectral graph convolution methods, etc., it can also provide more expressive graph neural networks for specific problems.
Important applications of graph neural networks: taking protein property prediction and sequence generation as an example
Next, I will share with you the application of graph neural networks in protein representation learning.Here I divide it into two categories: prediction models and generation models.
Protein feature encoding and property prediction
In terms of prediction tasks, we consider three types of tasks: mutant property prediction, solubility prediction, and subgraph matching, which are four specific tasks.
The first work is on mutation task prediction. As shown in the figure below, we used equivariant graph neural networks to characterize the internal spatial relationships of protein amino acids, where each node represents an amino acid, indicating the type, physical and chemical properties, and other characteristics of the amino acid at the point. The edge connections on the graph reflect the relationships between amino acids, such as the potential for common evolution and the influence of mutual forces.
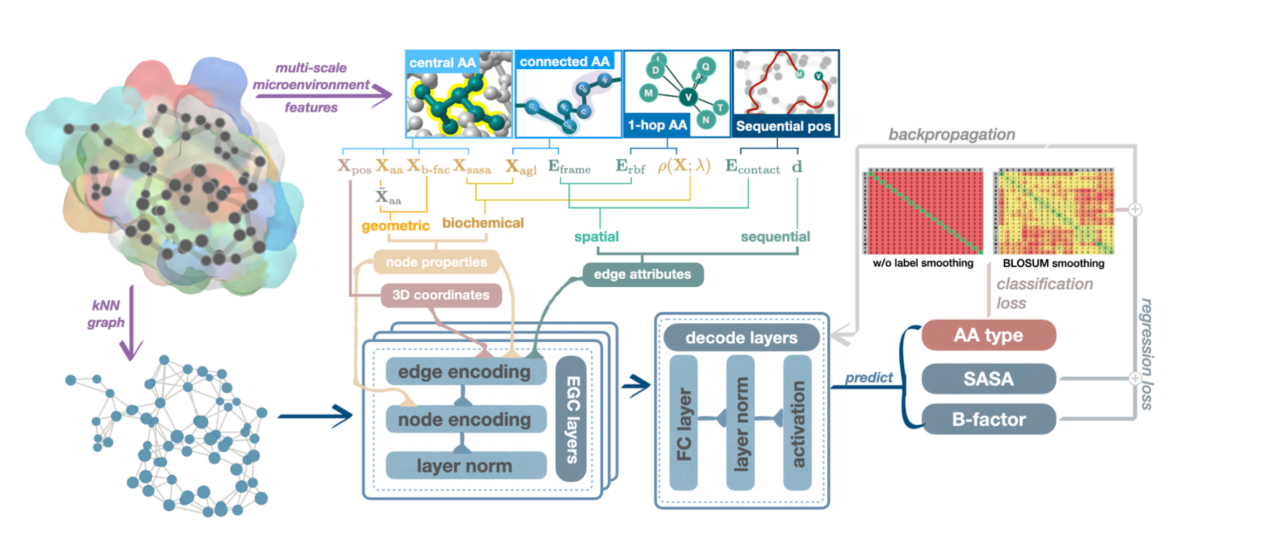
We then used a predictive model to score different mutants and identify high-scoring mutation combinations that are most likely to optimize protein properties. This lightweight graph neural network can significantly reduce training and data costs by integrating amino acids and the relationships between amino acids, while maintaining high performance while making the model small and beautiful. In addition, wet experimental verification on a variety of protein properties has proven that this model can significantly improve the effect and success rate of directed evolution. The study was published in ACS JCIM under the title "Protein Engineering with Lightweight Graph Denoising Neural Networks."
Paper address:
https://pubs.acs.org/doi/10.1021/acs.jcim.4c00036
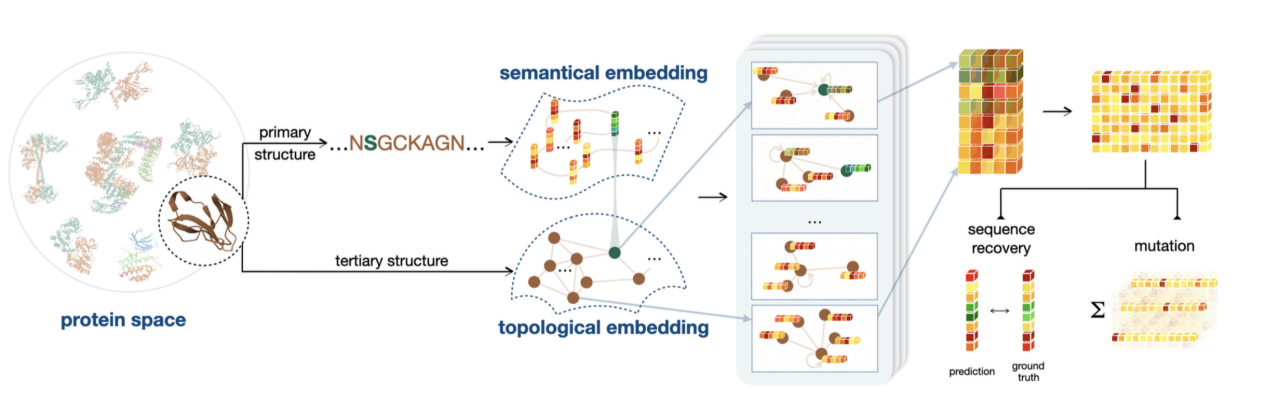
The second task is to further add protein sequence coding based on structural coding. This is because structural information assumes that the interactions between neighboring amino acids are stronger, while the interactions between distant amino acids are extremely weak. This assumption does not fully fit the actual situation, so sequence information is needed to supplement the consideration of long-range interactions. In addition, information on different biological properties has different emphases. For binding energy and thermal stability, structural information is dominant, but when it comes to properties such as catalytic activity, amino acid type information is more critical.
As shown in the figure below, we conducted experimental tests on more than 200 assays on ProteinGym and obtained the best performance of non-MSA methods. The study was published in eLife under the title "Semantical and Geometrical Protein Encoding Toward Enhanced Bioactivity and Thermostability".
Paper address:
https://elifesciences.org/reviewed-preprints/98033
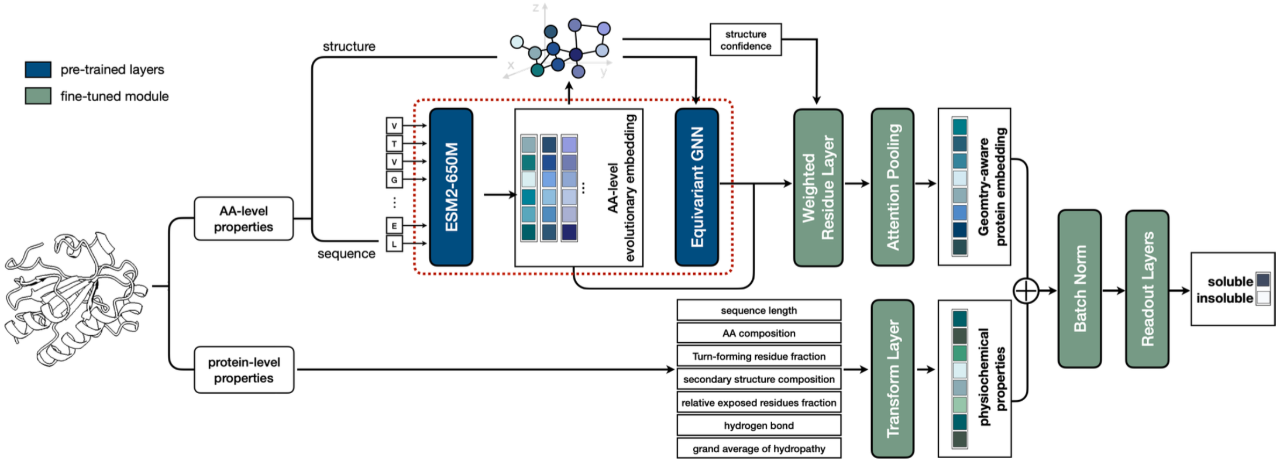
The amino acid level coding module of the third work is consistent with that of the second work. Information integration is based on protein sequence and structure. The difference is that it also integrates a variety of protein-level information based on prior knowledge, such as protein length, proportional distribution of 20 amino acids, etc.
As shown in the figure below, we tested the model's prediction effect on protein solubility and achieved SOTA results on thousands of test data based on calculations and experiments. The research is titled "ProtSolM: Protein Solubility Prediction with Multi-modal Features" and has been accepted by IEEE BIBM2024 (CCF Class B Conference).
Preprint paper address:
https://www.arxiv.org/abs/2406.19744
The fourth task is to explore the local similarities of protein structures. As shown in the figure below, although the protein is large as a whole, its core may lie in certain local structural features. In addition, from a macroscopic perspective, two proteins may be completely different at the sequence and structure levels, but have similar or even identical core functional modules.
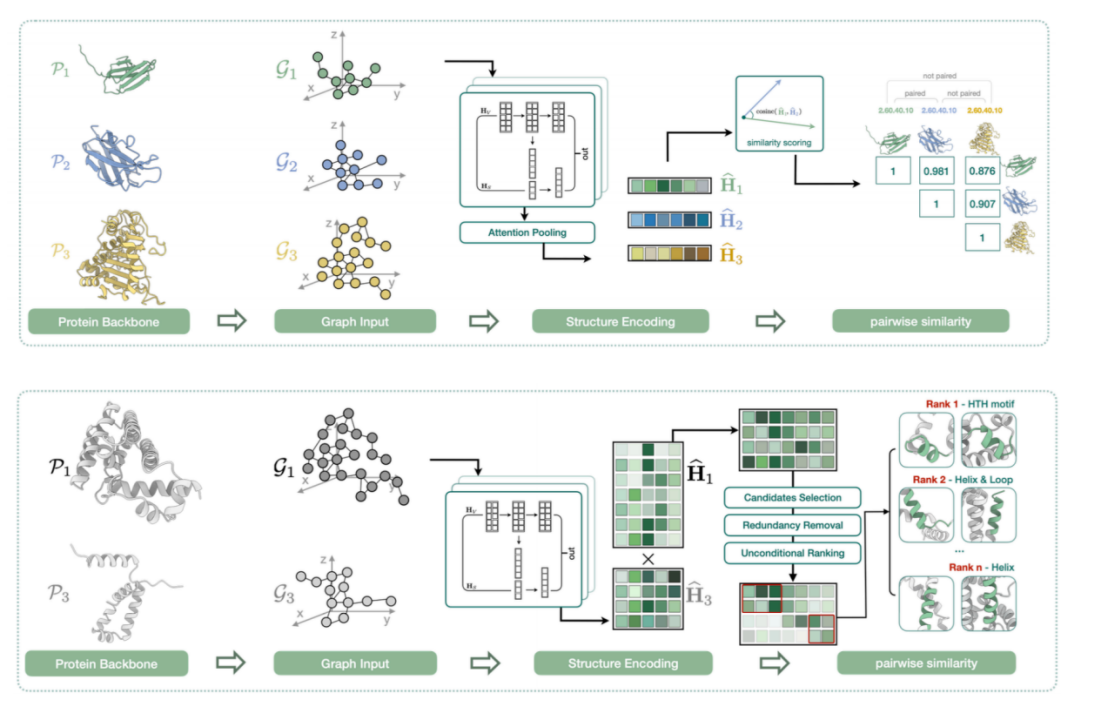
Therefore, we find an implicit expression for the local structure of each protein and calculate the similarity between these vectors. In addition to comparing the one-to-one similarity between structures, we also evaluate whether there are alignable local structure fragments between two complete proteins. The study is titled "Protein Representation Learning with Sequence Information Embedding: Does it Always Lead to a Better Performance?" and has been accepted by IEEE BIBM2024.
Preprint paper address:
https://arxiv.org/abs/2406.19755
Sequence Generation
Next, I will share with you two works, namely designing suitable amino acid sequences for protein structures. The core models of these two works are diffusion probability models (Diffusion).
The first task is to design a complete protein sequence based on the known amino acid skeleton to improve protein performance. The model framework is shown in the figure below. Unlike directed evolution, we modified hundreds of amino acids at a time to obtain a protein sequence with higher diversity. On the one hand, this method may find a new starting point for evolution and avoid the common problems of local optimality and negative up-regulation effects in directed evolution; on the other hand, by modifying more amino acids to obtain proteins with lower sequence similarity but the same function, it is possible to break the patent blockade.
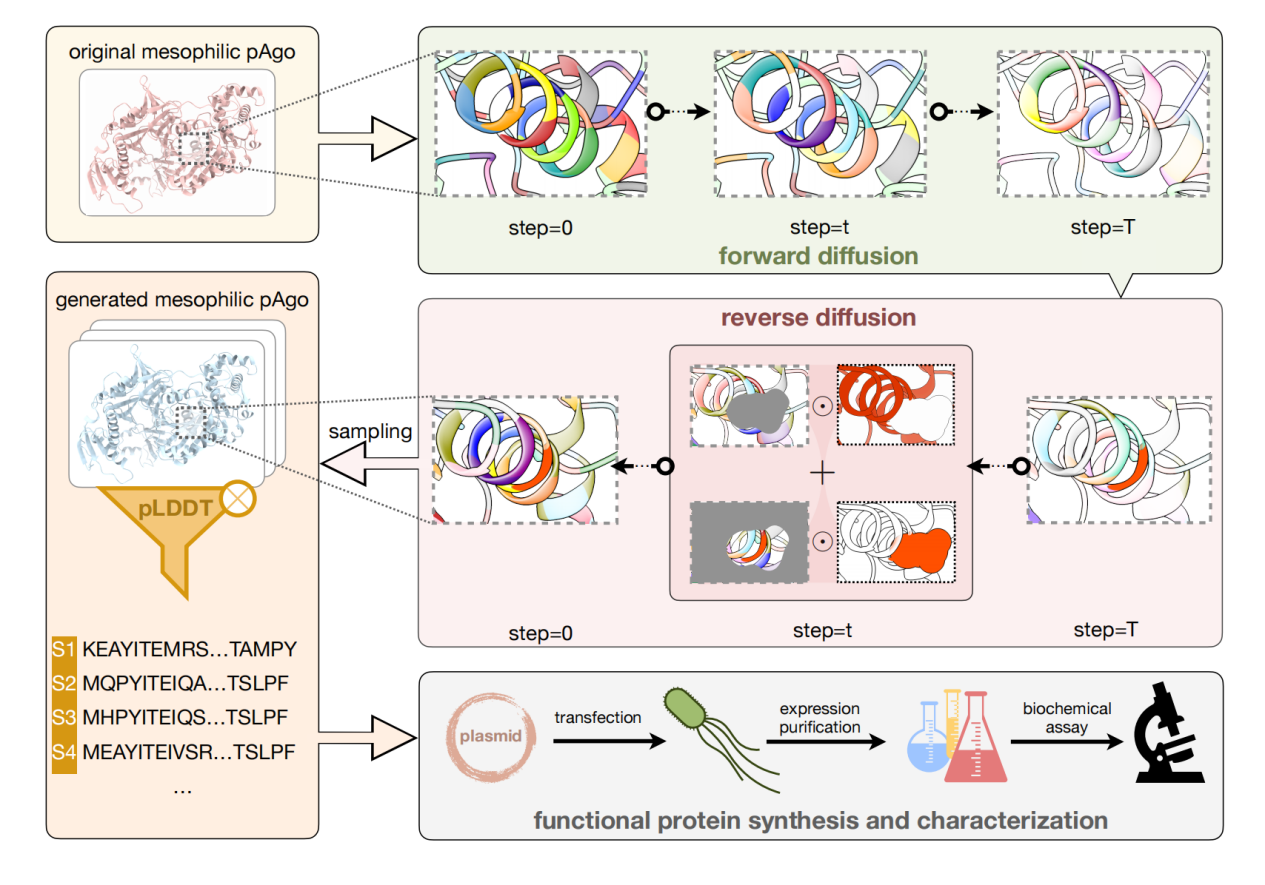
We used two Argonaute proteins (working at medium and ultra-high temperatures, respectively) as design templates, and generated more than 40 proteins that can mostly perform DNA cleavage at room temperature. The best design has a cleavage activity that is more than 10 times higher than the wild type, and its thermal stability is also significantly improved. The study was published in Cell Discovery under the title "Conditional Protein Denoising Diffusion Generates Programmable Endonucleases".
Preprint paper address:
https://www.biorxiv.org/content/10.1101/2023.08.10.552783v1
The second task, as shown in the figure below, is to independently determine the number and position of amino acids to be filled based on the secondary structure without strictly restricting the amino acid backbone structure. Compared with the skeleton-based generation method, this more coarse-grained generation condition can introduce sequence diversity into the generated sequence, and can also meet the specific needs of transforming and designing new proteins (for example, for transmembrane proteins, only the transmembrane part is constrained to be a helix structure, but the length and specific skeleton of this part are not strictly restricted). The study, titled "Secondary Structure-Guided Novel Protein Sequence Generation with Latent Graph Diffusion", was accepted by ICML AI4Science and the full text is under review.
Preprint paper address:
https://arxiv.org/html/2407.07443v1
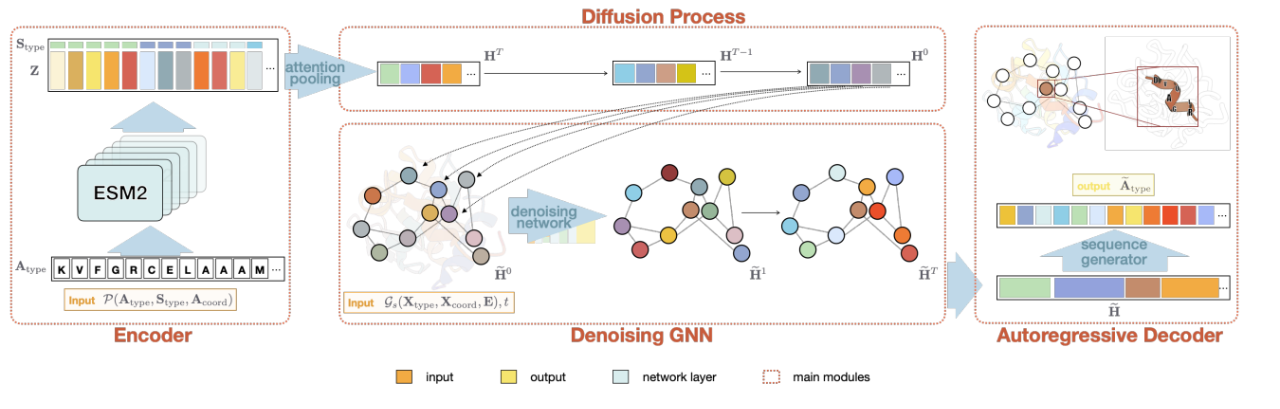
The above two protein sequence design works based on Diffusion can either generate a whole sequence according to the protein skeleton, or fix some key amino acids and skeleton structure and use them as generation conditions to fill in the amino acid sequence of the unfixed part.
Application of Graph Neural Networks to More Biological Problems
In addition to conventional molecular graph modeling, graph neural networks can also be applied to other types of data and problems to promote the research of more biological problems. Next, I will share two examples.
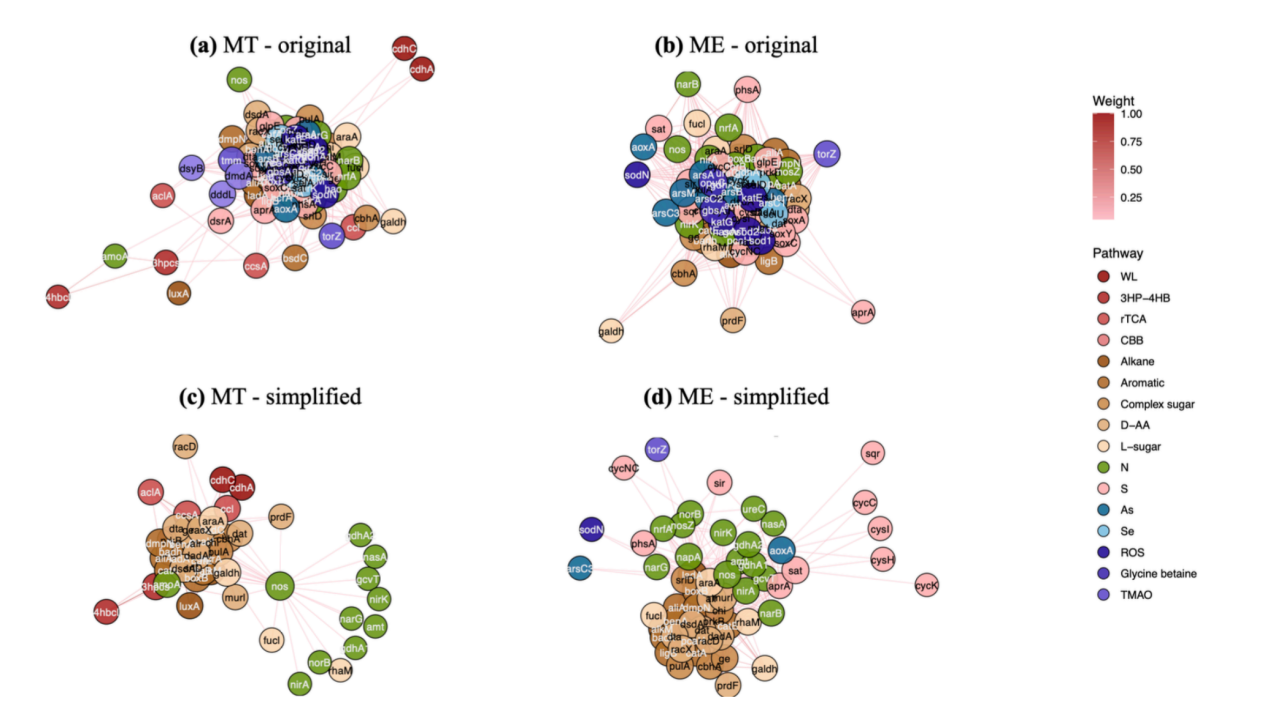
The first example is biological social network analysis and simplification. Similar to the complex relationships in human social networks, there is a lot of content worth exploring in biological social networks at various levels (such as microbial networks, gene networks, etc.).
We have used gene co-occurrence networks to simplify social networks. As shown in the figure below, Figures a and b are different networks of the same gene from the deep sea and the mountains. Their original forms are complex and disorganized. By building a graph neural network similar to the human social network, we simplified the two networks, identified the genes that occupy an absolute dominant position, and distinguished which genes have closer connections and which genes have relatively weaker connections. The simplified network can help biologists use their professional knowledge to analyze networks and biological communities. The preliminary version of the study is titled "A unified view on neural message passing with opinion dynamics for social networks."
Preprint paper address:
https://arxiv.org/abs/2310.01272
The second example is the interpretability research based on graph neural network. An intuitive example is that graph neural networks can help identify key local structures within molecules. On the one hand, this result can be used to test the rationality of the model. For example, when predicting protein function, if the model can locate key atoms or amino acids near the active center to a certain extent, it shows that the model has a certain rationality. On the contrary, if the model's attention is randomly and discretely distributed on multiple amino acids on the surface of the protein, the model may have problems. On the other hand, ideally, a reasonable and powerful explanatory model, by analyzing the role of each node in function prediction, may even help identify pocket areas of new proteins in the future.
Although large models have provided rich successful experience in many application scenarios, they are not the only solution to all problems. As a field where various structured data naturally exist, graph neural networks can provide possible solutions to many problems in biology. Whether it is molecules, complexes, genes, microbial networks, or larger and more complex systems, graph neural networks can provide a simple solution by maximizing human prior knowledge even with a small amount of data by implanting inductive biases.
About Zhou Bingxin

Zhou Bingxin is currently an assistant researcher at the National Center for Applied Mathematics (Shanghai Jiao Tong University Branch) at Shanghai Jiao Tong University. She received her Ph.D. from the University of Sydney, Australia in 2022, and later visited the University of Cambridge, UK as a visiting scholar. Her research focuses on using deep learning (especially geometric deep learning) to solve challenges in biology, such as enzyme engineering, metabolic gene networks, and protein structure group evolution analysis. The deep learning algorithms developed are used to process static, dynamic, heterogeneous, and noisy graphs, and some of them have been published in top international journals and conferences such as IEEE TPAMI, JMLR, ICML, and NeurIPS; the general deep learning framework for protein engineering and sequence design can effectively design and significantly improve the activity of complex proteins, and some of the results have been published in journals such as eLife, Chem. Sci., and ACS JCIM.
Personal homepage:
https://ins.sjtu.edu.cn/peoples/ZhouBingxin
Google Scholar:








