Command Palette
Search for a command to run...
A New Era of Material Exploration! Tsinghua University's Xu Yong and Duan Wenhui Team Released a Neural Network Density Functional Framework to Open the Black Box of Material Electronic Structure Prediction!

Density functional theory (DFT), as a key tool for predicting and explaining the properties of materials, plays an indispensable role in physics, chemistry, materials science and other fields.However, traditional DFT usually requires a lot of computing resources and time, and its application scope is significantly limited.
As an emerging interdisciplinary field, combining deep learning with density functional theory to predict and discover new materials through a large number of computational simulations is gradually overcoming the time-consuming and complex shortcomings of traditional DFT calculations, and has great application potential in the construction of computational material databases. Neural network algorithms accelerate the construction of larger-scale material databases, and larger data sets can train more powerful neural network models. However, most of the current deep learning DFT research handles DFT tasks and neural networks separately, which greatly limits the coordinated development between the two.
In order to combine the neural network algorithm and the DFT algorithm more organically, the research group of Xu Yong and Duan Wenhui from Tsinghua University proposed the neural-network density functional theory (neural-network DFT) framework.This framework unifies the minimization of loss functions in neural networks and the optimization of energy functionals in density functional theory. Compared with traditional supervised learning methods, it has higher accuracy and efficiency, and opens up a new path for the development of deep learning DFT methods.
The research was titled “Neural-network density functional theory based on variational energy minimization” and published in Phys. Rev. Lett.
Research highlights:
* This study proposes a theoretical framework for neural network density functional theory, combining variational density functional theory with equivalent neural network
* This study is based on the Julia language and combined with the Zygote automatic differentiation framework to develop a set of calculation programs called AI2DFT from scratch. In AI2DFT, automatic differentiation (AD) can be used for both variational DFT calculations and neural network training.
Paper address:
https://journals.aps.org/prl/abstract/10.1103/PhysRevLett.133.076401
The open source project "awesome-ai4s" brings together more than 100 AI4S paper interpretations and provides massive data sets and tools:
https://github.com/hyperai/awesome-ai4s
Deep learning and DFT integration to achieve unsupervised learning of physical information
Kohn-Sham DFT is the most widely used first-principles calculation method in material simulation.This method simplifies the complex electron interaction problem and maps it into a simplified non-interacting electron problem described by an effective single-particle Kohn-Sham Hamiltonian, and uses an approximate exchange-correlation function to consider complex multi-body effects. Although the Kohn-Sham equation is formally derived from the variational principle and is highly favored in the field of theoretical physics, it is not often used in DFT calculations because iterative solutions to differential equations are more efficient.
However, with the further integration of deep learning and DFT, this development paradigm has been completely changed.
The research of deep learning DFT mainly relies on data-driven supervised learning technology. As shown in Figure a below, in general, the traditional data-driven supervised learning method generates training data by performing DFT SCF calculations on different material structures, and then predicts data similar to DFT results by designing and training neural networks. In this process, the neural network is separated from DFT.
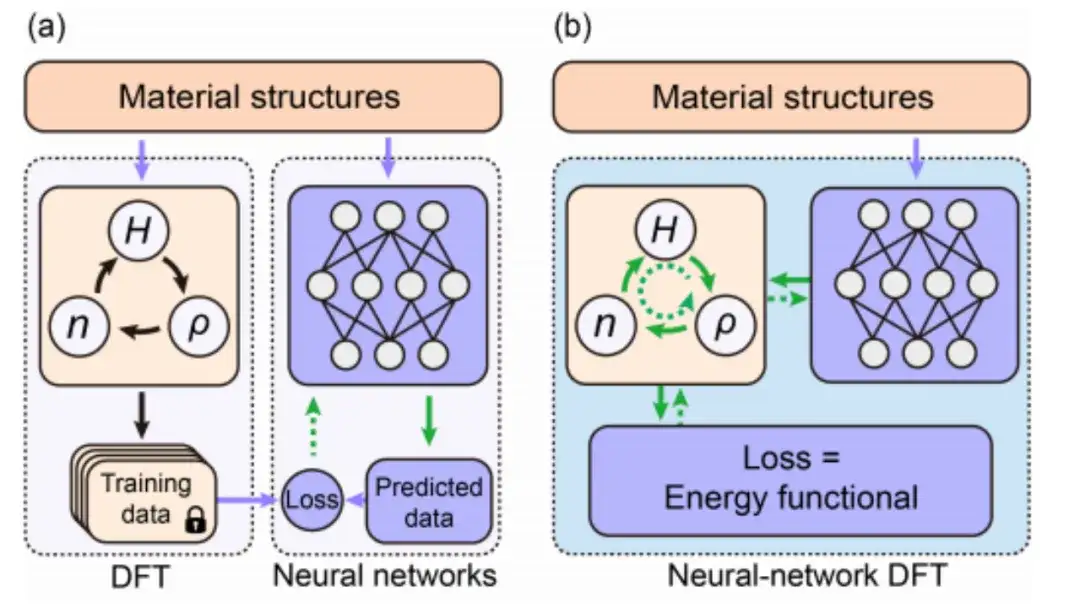
Nowadays, as shown in Figure b above, unsupervised learning of physical information based on neural network DFT realizes the sharing and collaboration of neural network algorithm and DFT algorithm by combining the optimization of energy functional in DFT with the minimization of loss function in neural network.More importantly, by explicitly introducing DFT into deep learning, the neural network model can simulate the real physical environment better than previous data training methods.
The introduction of automatic differentiation and back propagation techniques numerically implements the neural network DFT
This study selected the energy functional E[H] for follow-up study and adopted DeepH-E3 to ensure the covariance of the mapping {R} → H in the three-dimensional Euclidean group.
* DeepH-E3, the second generation DeepH architecture designed by Xu Yong's team based on equivalent neural network. This framework uses a covariant neural network under a three-dimensional Euclidean group to predict the density functional theory (DFT) Hamiltonian corresponding to the microscopic atomic structure, greatly accelerating the first-principles electronic structure calculation.
Specifically, as shown in the figure below, this study uses the embedding of material structure information as the input condition of the equivariant neural network, and then outputs the Hamiltonian matrix to obtain the neural network weight parameterized Hθ. The energy functional E[H] can also be regarded as the loss function E[Hθ] of the neural network.
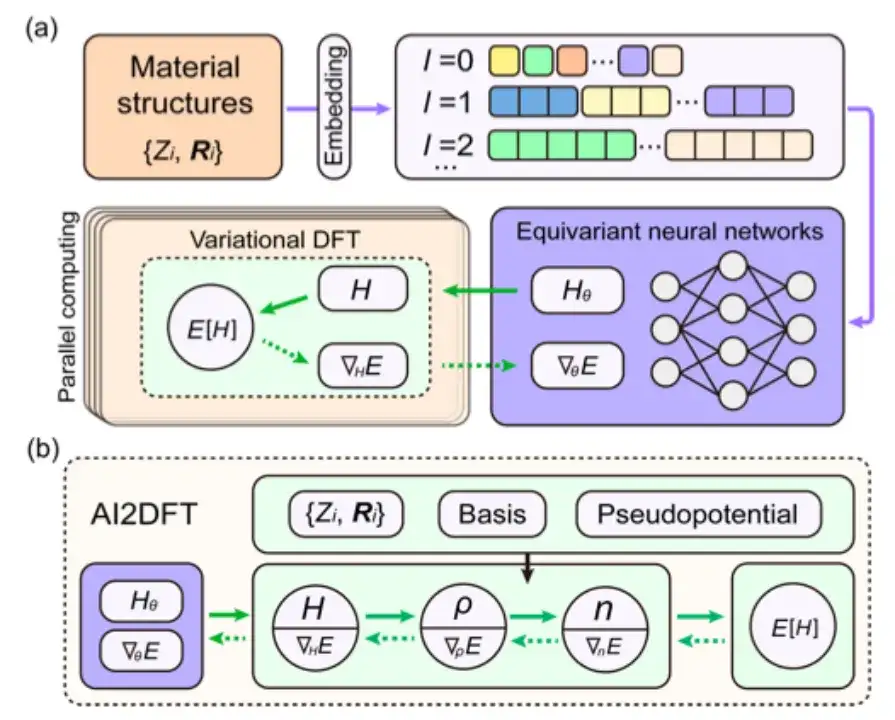
In neural network DFT, the DFT program must provide ∇HE to optimize the neural network parameters, which is a major challenge for DFT programming.
Although automatic differentiation (AD) is very suitable for calculating ∇HE, most current DFT codes do not fully support AD functions. Therefore, this study developed an autonomous and usable automatic differentiable DFT program "AI2DFT" using the Julia language. It is worth noting that in AI2DFT, automatic differentiation (AD) is applicable to both DFT calculations and neural network training.
In DFT, AI2DFT can deduce ρ and n from H to calculate the total energy, and then use reverse-mode AD to calculate ∇nE, ∇ρE, and ∇HE in turn based on the chain rule. In neural networks, the gradient information ∇θE can be used for neural network optimization.
Research results: Neural network DFT has high reliability and excellent performance in prediction accuracy
After establishing the theoretical framework of neural network DFT and completing the numerical implementation using the differentiable AI2DFT code, the study conducted comprehensive tests on a series of materials including H2O molecules, graphene, monolayer MoS2 and body-centered cubic Na.
Specifically, the study first checked whether the SCF iteration of AI2DFT could reproduce well the benchmark results of the SIESTA code, and then applied variational DFT to study the same materials.As shown in Figures a and b below, after dozens of variational steps, the total energy can converge below the μeV scale, and other physical quantities such as energy gradient, Hamiltonian, density matrix and charge density also show exponential convergence, which verifies the reliability and robustness of variational DFT.
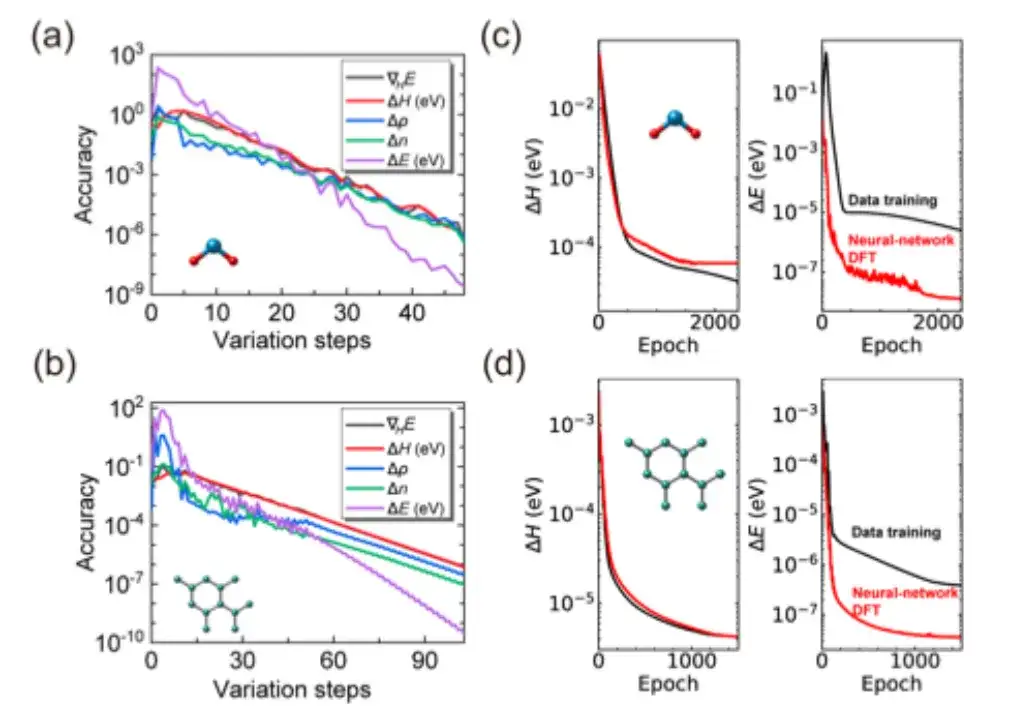
Moreover, as shown in Figures c and d above, compared with traditional data-driven supervised learning methods, AI2DFT combines the performance of variational DFT and DeepH-E3 neural network. For example:
* For the H2O molecule, the DFT Hamiltonian can be optimized by neural network DFT to achieve high accuracy, with an accuracy of 0.06 meV for neural network DFT and 0.02 meV for data training.
* For graphene, both methods achieved a higher accuracy of 0.004 meV. Therefore, the reliability of the neural network method was verified.
* In addition, in terms of energy prediction accuracy, the performance of neural network DFT is significantly better than data training. For example, for H2O molecules, the energy prediction accuracy of neural network DFT reaches 0.013 μeV, which is more than 60 times higher than the 0.83 μeV of data training.
Finally, the study also applied neural network DFT to the calculation of various material structures and demonstrated the ability of unsupervised learning.Taking the H2O molecule as an example, as shown in Figure a below, the study first obtained a pre-trained neural network model through data-driven supervised learning based on the DeepH-E3 method, and then used the neural network DFT to fine-tune 300 training structures to achieve high-precision predictions of the Hamiltonian and other physical quantities.
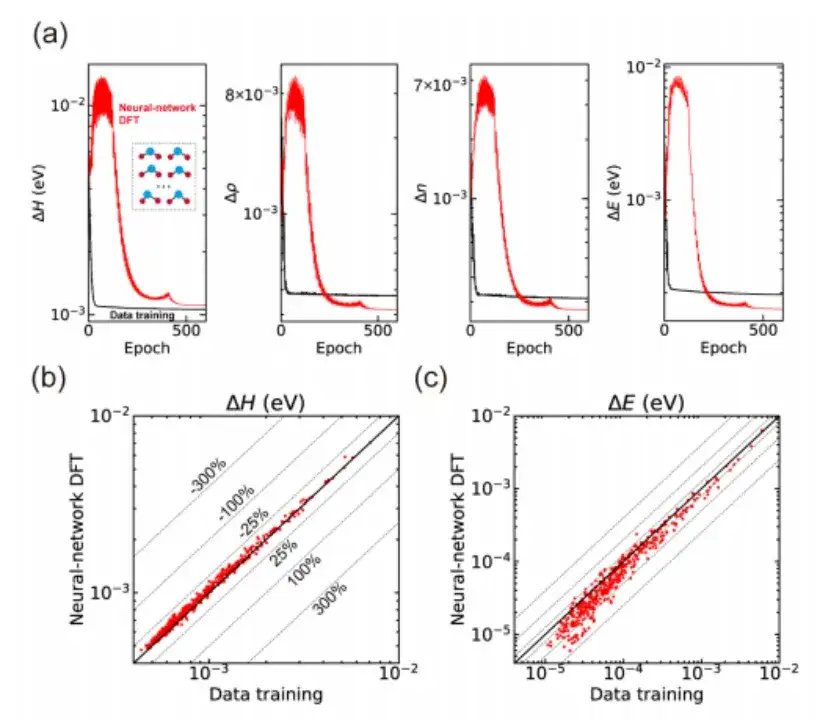
Not only that, the study also used the trained neural network model to predict 435 test structures that had not been seen during the training process, and the results also showed good generalization ability, as shown in Figures b and c above.Although the Hamiltonian produced by neural-network DFT has a slightly larger mean absolute error compared to data-driven supervised learning, it shows superior performance in terms of prediction accuracy, which implies that the neural network learns more physical patterns through the neural-network DFT process.
Focusing on DFT and first-principles calculations, AI will lead a new era of materials science
Led by Professor Xu Yong and Professor Duan Wenhui of Tsinghua University, the research team brought together Li Yang, Tang Zechen, Chen Zezhou and others. They have achieved a series of results in density functional theory (DFT) and first-principles calculations. Their work has promoted the widespread application of deep learning technology in materials science and physics research.
Since 2022, the research team of Xu Yong and Duan Wenhui has made major breakthroughs in the field of first principles computing.They developed a theoretical framework and algorithm based on deep learning - DeepH (Deep DFT Hamiltonian).This method makes full use of the locality principle of electronic properties. It only needs to use a model trained with a small system data set to give accurate predictions in large-scale material systems, significantly improving the computational efficiency of the electronic structure of non-magnetic materials.
The related results were published in Nature Computational Science under the title "Deep-learning density functional theory Hamiltonian for efficient ab initio electronic-structure calculation".
Paper link:
https://www.nature.com/articles/s43588-022-00265-6
In 2023, in order to further study the dependence of the DFT Hamiltonian of magnetic materials on atoms and magnetic structures, the research team of Xu Yong and Duan Wenhui proposed the xDeepH (extended DeepH) method.This method uses a deep equivariant neural network framework to represent the DFT Hamiltonian of magnetic materials, thereby performing efficient electronic structure calculations. This achievement not only provides an efficient and accurate calculation tool for the study of magnetic structures, but also proposes a feasible path for balancing the accuracy and efficiency of DFT.
The related results were published in Nature Computational Science under the title "Deep-learning electronic-structure calculation of magnetic superstructures".
Paper link:
https://www.nature.com/articles/s43588-023-00424-3
In order to design a neural network model that incorporates prior knowledge and symmetry requirements, the researchers further proposed the DeepH-E3 method.This method can use a small amount of DFT data to train a small-scale system and then quickly predict the electronic structure of a large-scale material system. It not only greatly improves the calculation speed by several orders of magnitude, but also achieves a high standard of sub-millielectronvolt in prediction accuracy.
The related results were published in Nature Communications under the title "General framework for E(3)-equivariant neural network representation of density functional theory Hamiltonian".
Paper link:
https://www.nature.com/articles/s41467-023-38468-8
With the continuous optimization of the theoretical framework, DeepH has achieved comprehensive coverage from non-magnetic to magnetic materials, and has also achieved significant improvements in prediction accuracy.In this context, the research team of Xu Yong and Duan Wenhui further used the DeepH method to construct a DeepH general material model.The model is able to cope with material systems containing multiple elements and complex atomic structures, and demonstrates excellent accuracy in predicting material properties.
The relevant research results were published in Science Bulletin under the title "Universal materials model of deep-learning density functional theory Hamiltonian".
Paper link:
https://doi.org/10.1016/j.scib.2024.06.011
Now, the research group of Xu Yong and Duan Wenhui has organically combined the neural network algorithm with the DFT algorithm, opening up a new path for the development of deep learning DFT methods. Undoubtedly, with the continuous advancement of deep neural network algorithms and the establishment of larger data sets, AI will become more intelligent, and it is likely that in the near future, first-principles calculations, material discovery, and design will all be performed by neural networks. This indicates that AI will lead materials science into a new data-driven era.








