Command Palette
Search for a command to run...
Selected for ICML! MIT Team Achieves New Breakthrough Based on AlphaFold, Revealing the Dynamic Diversity of Proteins
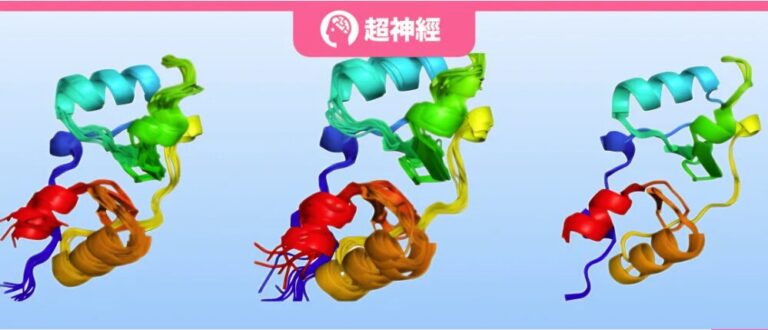
As an important component of organisms, proteins have different states and adopt complex three-dimensional structures based on different structural combinations of collective motion or disordered fluctuations to perform rich biological functions. For example, protein conformational changes are crucial to the functions of transporters, channels and enzymes, while the properties of the balanced combination help control the strength and selectivity of molecular interactions.
In recent years, deep learning methods such as AlphaFold have achieved great success in single-state protein modeling, but they cannot explain conformational heterogeneity. Therefore, for structural biologists,How to ensure accurate prediction of a single structure while revealing potential structural combinations?It is a difficult problem that needs to be solved urgently.
Recently, a research team from MIT combined the novel sampling methods of AlphaFold and ESMFold and provided a new perspective to observe and understand the conformational space of proteins through flow matching technology.
This study demonstrates the performance of flow matching variants AlphaFlow and ESMFlow in two different scenarios.The model was ultimately fine-tuned on the PDB and further trained on the ATLAS dataset, both showing excellent performance, not only surpassing the traditional MSA baseline in predicting conformational flexibility and atomic position distribution modeling, but also making significant progress in replicating high-order group observations.
The related research, titled "AlphaFold Meets Flow Matching for Generating Protein Ensembles", has been selected for ICML 2024, the top academic conference in the field of AI.

Paper address:
https://openreview.net/forum?id=rs8Sh2UASt
The open source project "awesome-ai4s" brings together more than 100 AI4S paper interpretations and provides massive data sets and tools:
https://github.com/hyperai/awesome-ai4s
Dataset: Based on PDB and ATLAS datasets to ensure the fairness of experimental results
As we all know, AlphaFold is developed and trained in an end-to-end manner based on structures in the PDB, while ESMFold uses embeddings from the Protein Language Model (PLM) as input.This study mainly used PDB dataset and MD dataset.
First, to construct a test set of structurally heterogeneous proteins from the PDB, the study used the SIFTS annotation database and its residue-level mapping from PDB chains to UniProt reference sequences to associate each deposited chain with a fragment. Subsequently, the study fully connected all fragments of the cluster based on a Jaccard similarity threshold of 0.75, treating each resulting cluster as a unique protein.This resulted in 75,000 proteins.
In addition, the study collected:
* Proteins that did not submit chains before the AlphaFold training deadline, but deposited 2-30 chains after the deadline;
* Proteins with lengths between 256–768 residues;
* Proteins with at least 2 structural clusters when the threshold for chain clustering was 0.85 symmetric lDDT-Cα and complete connectivity.
Finally, 563 proteins represented by 2,843 chains were obtained.The researchers extracted 100 proteins represented by 500 chains to form a test set.
Secondly, the researchers built the ATLAS dataset based on the MD dataset.The latter consisted of 1,390 proteins selected based on the ECOD domain classification.For each protein, the dataset provides 3 replicate simulations of 100 ns in length, each containing 10,000 frames. To train and validate on these trajectories, we first generated MSAs for all 1,390 ATLAS entries using the provided sequences and the ColabFold MMSeqs2 pipeline.
Subsequently, the researchers randomly selected 300 conformations from the training pipeline, using May 1, 2018 and May 1, 2019 as the training and validation deadlines, respectively, and finally obtained 1265/39/82 sets of training, validation, and test sets.

Model building: Using AlphaFold as a denoising model to perform flow matching on protein collections
Given the considerable challenges of redeveloping a distribution model with the same accuracy and generalization capabilities as AlphaFold, this study leverages recent conceptual advances in generative models.It is almost straightforward to reuse AlphaFold as a generative model.

To date, typical diffusion model architectures from text to image have almost all modeled the conditional distribution p(x | s) of an image x conditioned on a textual prompt s. At the core of these models is a denoising neural network that takes in a noisy image and a textual prompt and predicts a clean image.
Based on these conditions, such models are often trained with a simple mean squared error (MSE) objective. Similarly, a protein structure predictor trained with a regression-like loss function such as AlphaFold or ESMFold can be converted to a denoised model simply by providing additional noisy structure input. With these architectural adjustments, the study further allows AlphaFold and ESMFold to be plugged into any generative modeling framework based on iterative denoising.
The study believes that the design of the flow matching generation framework is equivalent to selecting a conditional probability path pt(x | x1) and its corresponding vector field ut(x | x1). Therefore, the study defines the conditional probability path by sampling noise x0 from q(x0) and linearly interpolating it with the data point x1, and then defines a reparameterized neural network x1(x, t; θ) ,Thus, the AlphaFold architecture is used as a denoising model.
To apply flow matching to protein structures, the study also describes the structure by the 3D coordinates of its β-carbons (α-carbon for glycine): x ∈ R^N×3. This also ensures that the input to the neural network is always a polymer-like, physically reasonable 3D structure.
Since the flow matching framework involves defining and reversing noise processes, it has many similarities with harmonic diffusion of protein structures, both of which converge to the same prior distribution. However, as a more general framework,Stream matching provides 2 main advantages:
first,Harmonic diffusion converges to the prior distribution only in the infinite time limit, and the convergence rate depends on the data dimension, i.e., protein size. This leads to distribution shift at inference time when training only on crops of relatively small size.
Secondly,Flow matching provides a simple way to handle missing residues that are very common in the PDB by simply omitting them. In contrast, harmonic diffusion creates dependencies between atomic positions and thus requires data interpolation for missing residues.

Finally, the study fine-tuned all the weights of AlphaFold and ESMFold on PDB based on the process matching framework, using AlphaFold and ESMFold with training deadlines of May 1, 2018 and May 1, 2020, respectively. At the end of this phase of training, the study obtained flow matching variants of AlphaFold and ESMFold,And called it AlphaFLOW and ESMFLOW.
To evaluate the ability to learn from MD ensembles, the study further fine-tuned the two models on the ATLAS dataset containing all-atom MD simulations. After training with 43,000 and 27,000 additional examples, respectively,The study obtained MD-specific model variants - AlphaFLOW-MD and ESMFLOW-MD.
Experimental results: Performance exceeds that of traditional methods and has broad application prospects in the field of structural biology
The researchers first evaluated the capabilities of AlphaFLOW and ESMFLOW for diverse conformations of proteins deposited in the PDB.
To this end, the study constructed a test set containing 100 proteins with evidence of multiple chains and conformational heterogeneity deposited after the AlphaFold training deadline (May 1, 2018), and evaluated them for three major indicators: precision, recall, and diversity.

The results show that AlphaFLOW is similar to MSA subsampling in that both increase the diversity of predictions at the expense of accuracy, but compared to MSA subsampling, AlphaFLOW variants track significantly better Pareto fronts.
In terms of precision and recall,AlphaFLOW exhibits very similar behavior to MSA subsampling.Somewhat surprisingly, neither approach significantly improves overall recall relative to the baseline AlphaFold.
Overall, ESMFold and ESMFLOW have relatively lower accuracy compared to the AlphaFold family of methods. However, ESMFLOW is able to inject a lot of diversity relative to the baseline ESMFold.And improve recall with almost no sacrifice in precision.
In addition, the RMWD analysis of this study showed that AlphaFlow was slightly better than AlphaFold in predicting the average atomic positions and significantly better than MSA subsampling in terms of modeling variance.

This study further evaluated the ability of AlphaFLOW and ESMFLOW to generate proxy MD ensembles for a test set of 82 proteins from the ATLAS database. The study sampled separately using each method and checked the similarity of the sampled samples to the MD population through a series of evaluations.
The results show thatAlphaFLOW-MD achieves significant improvements in similarity, far exceeding the performance of MSA subsampling.

Since MD is considered as true value but is expensive to run to convergence, we further analyzed whether AlphaFLOW can provide better results under equivalent limited computational budget, such as GPU hours. To this end, we reduced the number of samples drawn from AlphaFLOW (from 250 to 4) and shortened the length of MD trajectory (from 100ns to 160ps).
The results show that the quality of the AlphaFLOW ensemble remains constant, but the MD trajectories take longer to reach or exceed the same quality level.
Three major protein general pre-training models stand out, and the field of structural biology is full of vitality
In the past few years, proteins and AI have continuously collided to create new sparks.At present, the universal pre-training of proteins has formed a new situation of three pillars.That is, the DeepMind Alphafold series, David Baker's RoseTTAFold series, and the Meta ESM series. Based on these three models, related scientific research results have begun to explode. In the first half of 2024 alone, many research results were published in top journals such as Nature and Science.
In March 2024, researchers from the University of North Carolina School of Medicine, the University of California, San Francisco, Stanford University, and Harvard University published a study in Science confirming thatAlphaFold2 predicted structures can guide future drug discovery.The research team found that AlphaFold2 showed significant practicality in structural biology, protein design, interactions, target prediction, function prediction, and biological mechanisms, and was able to search for potential new drugs by screening billions of compounds and matching libraries with protein structures.
In May 2024, the Google DeepMind team released AlphaFold 3 in Nature, which expanded the technology beyond protein folding and can accurately predict the structure and interaction of life molecules such as proteins, DNA, RNA, ligands, etc. with unprecedented accuracy. This means thatAlphaFold 3 will further accelerate drug design and genomic research,Ushering in a new era of artificial intelligence cell biology.
With the release of AlphaFold 3,The Alphafold series has finally built an all-atomic foundation.Similarly, the RoseTTAFold series also successfully launched RoseTTAFold All-Atom in the first half of this year, realizing the ability to make reasonable predictions on protein covalent modifications and the assembly of multiple nucleic acid chains and small molecules.
With the help of Alphafold3 and RoseTTAFold All-Atom, researchers are giving full play to their imagination. For example, in June 2024, an international research team published a paper in Nature Biotechnology, showing how to use the strategy of combining AlphaFold 3 and RoseTTAFold All-Atom to successfully design a new type of protein scaffold that can more effectively deliver drugs directly to diseased cells, thereby improving treatment effects and reducing side effects. This discovery marks a solid step forward in the application of AI in precision medicine.
Unfortunately, in August 2023, Meta disbanded the ESMFold team and turned to fully promote the commercialization of AI. However, research on the ESM series has not stopped. For example, the model has made important progress in the field of protein language modeling and provided a unified modeling solution that integrates multi-scale information. It is worth noting that it is the first protein pre-trained language model that can process both amino acid information and atomic information.
It can be seen from this thatIn the new era where the Alphafold series, RoseTTAFold series, and ESM series are on par with each other,The combination of AI and protein research will become closer, which will not only accelerate our understanding of protein structure and function, but also bring revolutionary changes to disease treatment, drug development and biotechnology applications. With the leapfrog development brought about by AI technology, the field of structural biology is becoming more vibrant, and a new chapter in the field of biomedicine is slowly unfolding.








