Command Palette
Search for a command to run...
Selected for ICML! The Renmin University Team Used Equivariant Graph Neural Network to Predict Target Protein Binding Sites, With the Highest Performance Improvement of 20%
In living systems, almost all biological and pharmacological processes involve interactions between receptors (target proteins) and ligands (small molecules). These interactions occur in specific regions of the target protein structure.Known as "binding sites" - predicting the binding sites of target proteins plays a fundamental role in various downstream tasks such as drug discovery.
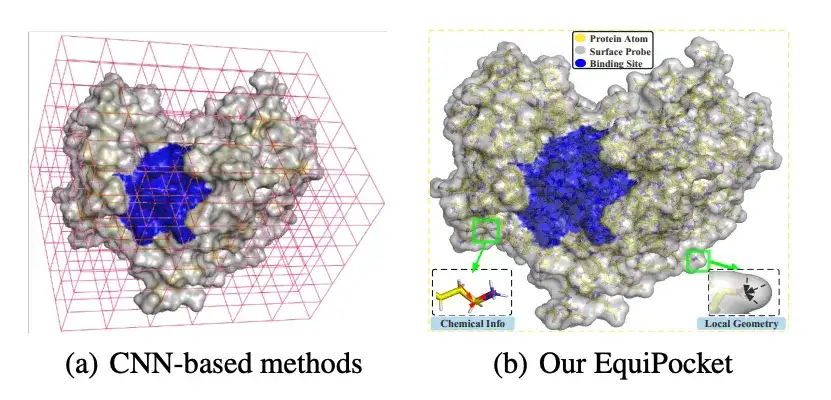
In recent years, inspired by the breakthrough of deep learning, convolutional neural networks (CNN) have been successfully applied to ligand binding site prediction. CNN-based methods cluster the atomic space of proteins into the nearest voxels, treat proteins as three-dimensional images, and then model binding site prediction as a target detection problem or semantic segmentation task on a 3D grid. These methods have certain advantages, but there are still challenges, such asIt has defects in representing irregular protein structures; it is sensitive to rotation; it does not adequately describe the geometric features of protein surfaces; and it is insensitive to changes in protein size.
To this end, a research team from the Gaoling School of Artificial Intelligence at Renmin University of China recently published a research paper titled "EquiPocket: an E(3)-Equivariant Geometric Graph Neural Network for Ligand Binding Site Prediction" at ICML 2024, the top academic conference in the field of AI. This study is the first to apply the E(3) equivariant graph neural network (GNN) to ligand binding site prediction.Proposed a framework called EquiPocket,The challenges encountered by CNN-based methods are addressed.
Research highlights:
* The first application of E(3) isovariant GNN to ligand binding site prediction
* Compared with traditional CNN-based methods, EquiPocket does not require voxelization, can model irregular protein structures, and is insensitive to any Euclidean transformation, thus solving challenges such as "defects in representing irregular protein structures" and "sensitivity to rotation"
* Extensive experiments on representative benchmark methods demonstrate the superiority of EquiPocket over the current state-of-the-art methods, which can help various downstream tasks such as drug discovery
Paper address:
https://openreview.net/forum?id=1vGN3CSxVs
The open source project "awesome-ai4s" brings together more than 100 AI4S paper interpretations and provides massive data sets and tools:
https://github.com/hyperai/awesome-ai4s
Dataset: Comprehensive verification of multiple professional datasets
The researchers selected several specialized datasets and evaluated them using a subset of mlig from each dataset that contained relevant ligands for binding site prediction.
in,scPDB is a well-known dataset for binding site prediction.Contains protein, ligand, and 3D cavity structures generated by VolSite. This study used the 2017 release for training and cross-validation, which contains 17,594 structures, 16,034 entries, 4,782 proteins, and 6,326 ligands.
PDBbind is a commonly used dataset for studying protein-ligand complexes.Contains three-dimensional structures of proteins, ligands, binding sites, and accurate binding affinity results determined in the laboratory. This study used the 2020 version for evaluation, which consists of two parts: the general set (14,127 complexes) and the refined set (5,316 complexes). The general set contains all protein-ligand complexes, and the refined set selects better quality compounds from the general set for experimental testing.
COACH 420 and HOLO4K are two test datasets used for binding site prediction.First introduced by (Krivák & Hoksza, 2018).
Model architecture: EquiPocket's overall framework consists of three modules
The overall framework of EquiPocket consists of 3 modules:As shown in the following figure:
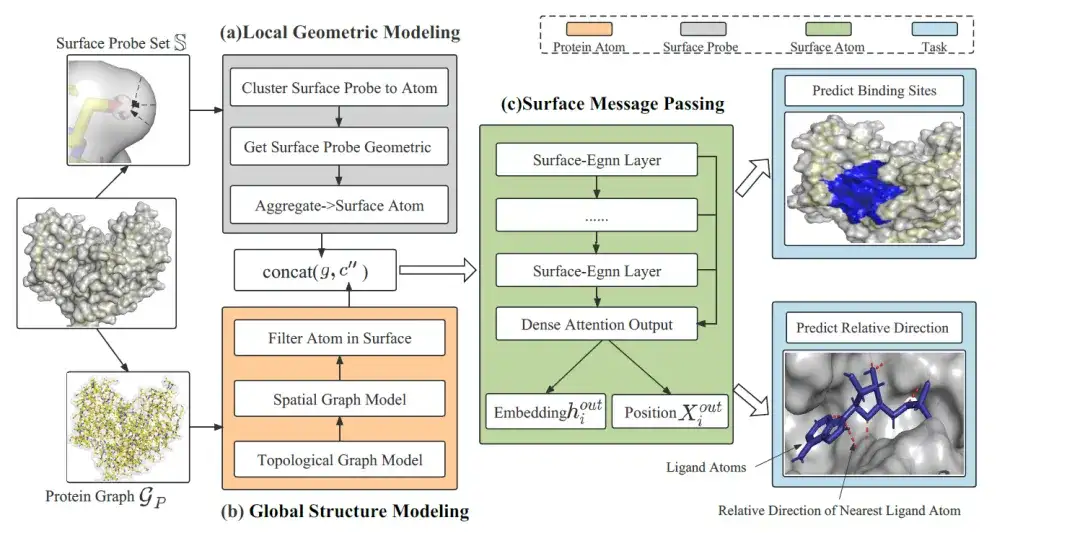
The first module is the Local Geometric Modeling module, which is used to extract the local geometric information of each surface atom; the second module is the Global Structure Modeling module, which is used to describe the chemical and spatial structure of proteins; the last module is the Surface Message Passing module, which captures the surface geometry by transmitting equivariant information on surface atoms.
Local geometry modeling module
The local geometry of each protein atom determines whether its nearby region is suitable to be part of the binding site.
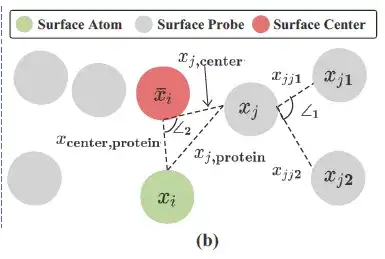
As shown in the figure above, researchers use surface probes (gray in the figure above) around each protein surface atom (Surface Atom, green in the figure above) to describe local geometric information. Specifically, for each surface atom i ∈ VS, the surface probes around it are returned by a subset of S, namely:
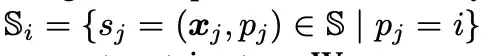
The researchers constructed geometric information based on Si and recorded the center/average value of all 3D coordinates in Si (Surface Cente, red in the figure above) as xi¯.
Global Structural Modeling Module
Although binding sites are primarily composed of surface atoms, the overall structure of the protein often affects ligand interactions as well as binding site formation and therefore needs to be modeled.
The researchers achieved this goal through two connected processes: chemical graph modeling and spatial graph modeling. The resulting global structural modeling module is responsible for processing information about the entire protein, including atom types, chemical bonds, relative spatial positions, etc.
Surface Information Transfer Module
Given the local geometric features of surface atoms and the global encoding features, this module will perform equivariant information transfer on the surface map to update all the features of the protein surface atoms.
Research results: EquiPocket improves performance by 10-20% compared to baseline models
In the experiment, the researchers chose to compare EquiPocket with the following baseline models:
* Geometry-based methods: Fpocket
* Machine learning method: P2rank
* CNN-based methods: DeepSite, Kalasanty, DeepSurf, RecurPocket
* Topology-based models: GAT, GCN and GCN2
* Spatial graph-based models: SchNet, EGNN
The indicators used to evaluate the model include DCC (the distance between the predicted binding site center and the actual binding site center), DCA (the shortest distance between the predicted binding site center and any ligand grid) and failure rate (the sampling rate without any predicted binding site center). The following table shows the results of binding site prediction on COACH 420, HOLO4K and PDBbind.
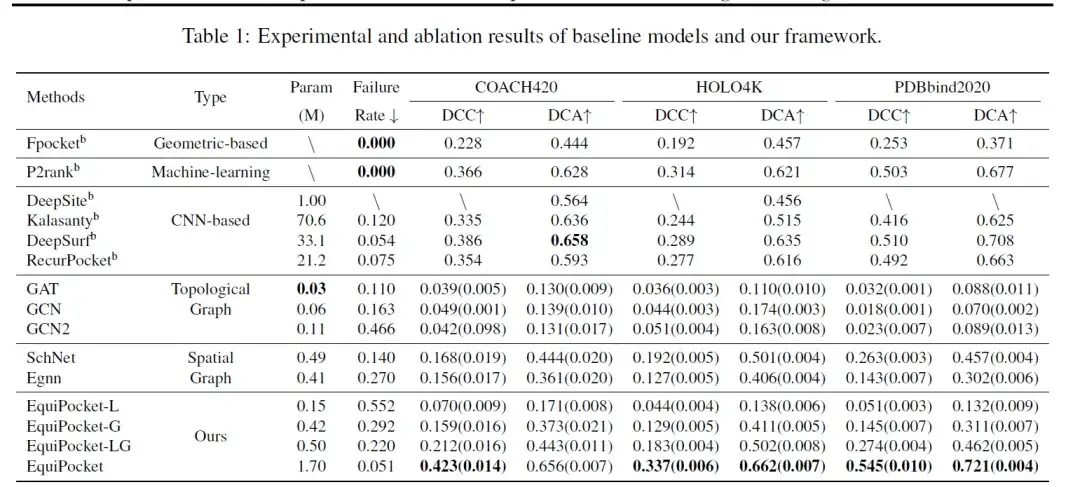
As shown in the data in the table,The geometry-based method Fpocket has poor performance.Since this method only uses the geometric features of the protein, the failure rate is 0; the machine learning method P2rank combines random forests with the geometric information of the protein surface, thereby significantly improving the performance.
CNN-based methods (DeepSite, Kalasanty, DeepSurf, RecurPocket) perform much better than geometry-based methods.Among them, DCC and DCA improve more than 50%, but require a large number of parameters and computing resources. Among them, the earlier proposed methods DeepSite and Kalasanty are limited by the size variation of proteins and the lack of ability to handle large proteins, which may lead to prediction failure.
For graph models,The performance of topological graph models (GCN, GAT, GCN2) is poor.This is mainly because they only consider atoms and chemical bonds, ignoring the spatial structure of proteins; spatial graph models (SchNet, EGNN) generally outperform topological graph models. EGNN uses the properties of atoms and their relative/absolute spatial positions, which works better; SchNet only updates the embedding based on the relative distance of atoms, but the performance of spatial graph models is worse than CNN-based and geometry-based methods because the former cannot obtain enough geometric features and cannot solve the problem of protein size changes.
The above results show thatThe geometric information and multi-level structural information of the protein surface are crucial for binding site prediction.
In addition, this also reflects the limitations of current GNN models, that is, it is difficult to collect enough geometric information from the protein surface or the required computing resources are too large to be applied to macromolecular systems such as proteins. Therefore, the EquiPocket framework can not only update chemical and spatial information at the atomic level, but also effectively collect geometric information without excessive computing resources.Its performance improves upon previous results by 10-20%.
From small molecule ligands to biological macromolecules, AI deeply interprets protein structure
There are billions of molecular machines inside every plant, animal and human cell, made up of molecules such as proteins, nucleic acids, sugars, and no single part of them can function alone - only by understanding how they interact in millions of combinations can we gain a deeper understanding of life.
In May this year, Google DeepMind released the AlphaFold3 model, which can predict the joint structure of complexes including proteins, nucleic acids, small molecules, ions and modified residues. The interaction between proteins and small molecule ligands is the core of the drug mechanism. AlphaFold3 can accurately predict the three-dimensional structure of protein-ligand binding through its advanced deep learning algorithm, with an accuracy far exceeding that of existing docking tools.
In terms of new drug development,Through the protein-ligand structures predicted by AlphaFold3, researchers can more effectively screen and design new drug candidates and accelerate the drug discovery process. In terms of optimizing existing drugs, this tool can also be used to optimize existing drugs by improving their binding mode with the target protein to enhance efficacy or reduce side effects.
In addition to small molecule ligands,Proteins also need to combine with biological macromolecules such as DNA and sugars to exert their biological functions.Currently, thousands of protein structure complexes have been deposited in the protein database through experimental methods. However, traditional experimental methods are time-consuming and expensive, while prediction methods based on machine learning can easily solve the challenges.
In February this year, a research team from Nanjing Agricultural University published an online research paper titled "ULDNA: Integrating Unsupervised Multi-Source Language Models with LSTM-Attention Network for High-Accuracy Protein-DNA Binding Site Prediction" in Briefings in Bioinformatics, an important journal in the field of biology.A new deep learning prediction method ULDNA was developed to predict protein-DNA binding sites.
Paper address:
https://academic.oup.com/bib/article/25/2/bbae040/7606634

The core idea of ULDNA is to use the protein language model to design feature representation for sequences, and then combine it with the long short-term memory network (LSTM-Attention Network) of the attention mechanism to train the DNA binding site prediction model. The researchers selected seven benchmark datasets (the number of protein sequences ranged from 40 to 600) such as PDNA-128, PDNA-316, and PDNA-335, and conducted a comprehensive test on ULDNA. The experimental results show thatULDNA performs well on all datasets, and its prediction performance is significantly better than that of the other nine mainstream methods.
In addition to DNA, sugars are ubiquitous on the cell surfaces of all organisms, where they interact with multiple protein families such as lectins, antibodies, enzymes, and transporters to regulate key biological processes such as immune responses, cell differentiation, and neural development.Understanding the interaction mechanism between carbohydrates and proteins is the basis for developing carbohydrate drugs.However, the diversity and complexity of carbohydrate structures, especially the variability of their protein binding sites, pose challenges to the acquisition of experimental data and drug design.
Recently, a team from the Chinese Academy of Sciences developed a deep learning model, DeepGlycanSite, which can accurately predict sugar binding sites on a given protein structure. DeepGlycanSite incorporates the geometric and evolutionary characteristics of proteins into a deep equivariant graph neural network based on the Transformer architecture.Its performance significantly surpasses previous advanced methods and can effectively predict the binding sites of various sugar molecules.
Combined with mutagenesis studies, DeepGlycanSite revealed guanosine-5'-diphosphate sugar recognition sites for important G protein-coupled receptors. These findings suggest that DeepGlycanSite is valuable for sugar binding site prediction and can provide insights into the molecular mechanisms behind carbohydrate regulation of therapeutically important proteins.

The study, titled "Highly accurate carbohydrate-binding site prediction with DeepGlycanSite," was published in Nature Communications on June 17, 2024.
Paper address:
https://www.nature.com/articles/s41467-024-49516-2
In short, proteins are important molecules in life and play a key role in the structure and function of cells. Studying the structure of proteins is of great significance for understanding life processes, revealing disease mechanisms, and drug development. Today, machine learning has opened a new door for scientists to understand the mysteries of life.
References:
1.https://openreview.net/forum?id=1vGN3CSxVs
2.https://mp.weixin.qq.com/s/aGzcr0ncQA-jBy-vTGC35Q
3.https://www.jiqizhixin.com/articles/2024-05-09
4.https://news.njau.edu.cn/2024/0








