Command Palette
Search for a command to run...
Neural Network Replaces Density Functional Theory! Tsinghua Research Group Releases Universal Material Model DeepH, Achieving ultra-accurate Prediction

In material design, understanding its electronic structure and properties is the key to predicting material properties, discovering new materials, and optimizing material properties.Density functional theory (DFT) is widely used in the industry to study the electronic structure and properties of materials. Its essence is to use electron density as the carrier of all information in the ground state of molecules (atoms).Instead of the wave function of a single electron, the multi-electron system is converted into a single-electron problem for solution, which not only simplifies the calculation process, but also ensures the calculation accuracy and can more accurately reflect the aperture distribution.
However, DFT is computationally expensive and can usually only be used to study small-scale material systems. Inspired by the Materials Genome Initiative, scientists have begun to try to use DFT to build a huge material database. Although only a limited data set has been collected so far, it is already a remarkable start. With this as a starting point, with the new changes brought about by AI technology, researchers began to think, "Can combining deep learning with DFT and allowing neural networks to deeply learn the essence of DFT bring about a revolutionary breakthrough?"
This is the core of the Deep Learning Density Functional Theory Hamiltonian (DeepH) method.By encapsulating the complexity of the DFT in a neural network, DeepH is not only able to perform calculations with unprecedented speed and efficiency, but its intelligence also continues to improve as more training data is added.Recently, the research group of Xu Yong and Duan Wenhui from the Department of Physics at Tsinghua University successfully used their original DeepH method to develop the DeepH universal material model and demonstrated a feasible solution for building a "big material model". This breakthrough provides new opportunities for innovative material discovery.
The related research, titled "Universal materials model of deep-learning density functional theory Hamiltonian", has been published in Science Bulletin.

Paper address:
https://doi.org/10.1016/j.scib.2024.06.011
The open source project "awesome-ai4s" brings together more than 100 AI4S paper interpretations and provides massive data sets and tools:
https://github.com/hyperai/awesome-ai4s
Build a large material database through AiiDA to eliminate magnetic material interference
To demonstrate the universality of the DeepH general material model, this study constructed a large material database containing 104 solid materials through the Automatic Interactive Infrastructure and Database (AiiDA).
To showcase the diverse elemental composition, the study also selected the first four rows of the periodic table, excluding transition elements from Sc to Ni to avoid interference from magnetic materials, and excluding rare gas elements. The candidate material structures are derived from the Materials Project database. In addition to filtering based on element type, candidate materials are further refined in the Materials Project to include only those marked as "non-magnetic." For simplicity, structures containing more than 150 atoms in the unit cell are excluded.
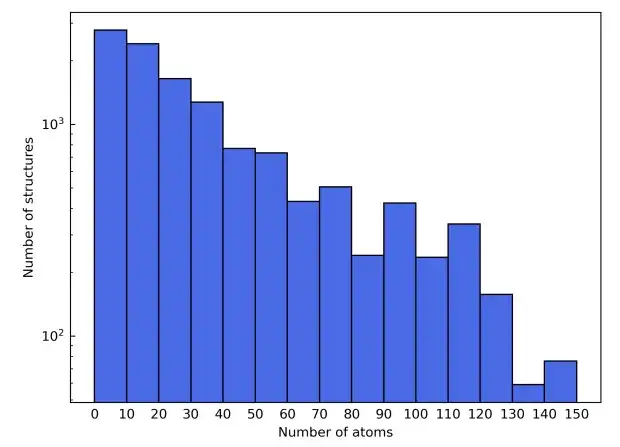
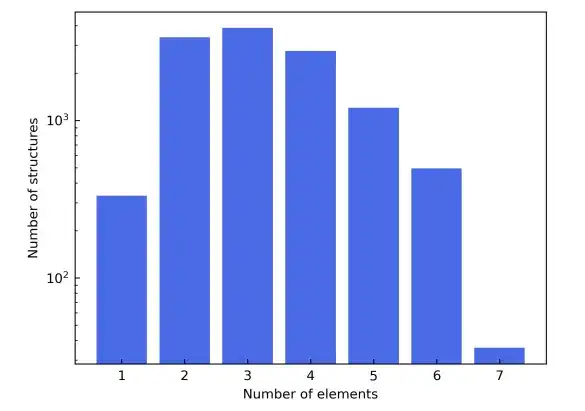
As a result of these filtering criteria, the final materials dataset consisted of a total of 12,062 structures. During training, the dataset was split into training, validation, and test sets in a 6:2:2 ratio. Next,The study used the AiiDA (Automated Interactive Infrastructure and Database) framework to develop a high-throughput workflow for density functional theory calculations and used it to build a materials database.
Using the DFT Hamiltonian as the target, DeepH is trained using the DeepH-2 method.
Research suggests thatThe DFT Hamiltonian is an ideal target for machine learning.
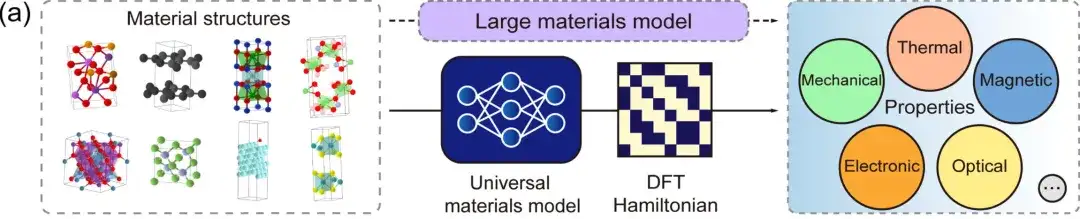
first,The DFT Hamiltonian is a fundamental quantity that can be directly derived from physical quantities such as total energy, charge density, band structure, and physical responses.The DeepH general material model can accept any material structure as input and generate the corresponding DFT Hamiltonian, so that various material properties can be directly derived, as shown in the figure above.
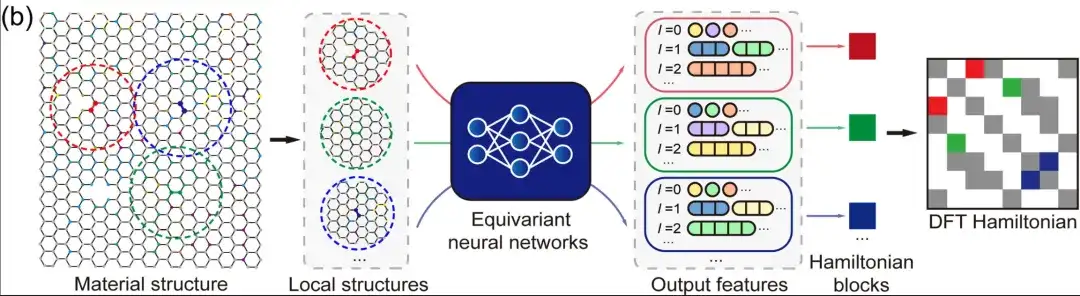
Secondly,In the case of a localized atomic basis set, the DFT Hamiltonian can be represented as a sparse matrix whose elements are determined by the local chemical environment.In equivariant neural networks, DeepH uses output features labeled with different angular quantum numbers l to represent the DFT Hamiltonian, as shown in the figure above. Therefore, people can model the Hamiltonian matrix elements between atomic pairs based on the adjacent structural information without having to model the DFT Hamiltonian matrix of the entire material structure. This not only greatly simplifies the deep learning task, but also greatly increases the amount of training data. In terms of reasoning, once the deep learning network learns enough training data, the trained model can be well generalized to more new material structures that have not been seen.
The key idea of DeepH is to use neural networks to represent HDFT.By varying the input material structure, HDFT training data generated by the DFT code is first created, which is then used to train the neural network. These trained network models are then used to infer new material structures.
In this process, there are two very important prior knowledge: one is the principle of locality,The study represents the DFT Hamiltonian in a localized atomic sample and decomposes the Hamiltonian into blocks that describe interatomic coupling or intraatomic coupling. Therefore, a single training material structure may correspond to a Hamiltonian block of a large amount of data. In addition, each Hamiltonian block can be determined based on information about the local structure rather than the entire structure. This simplification ensures the high accuracy and transferability of the DeepH model.
The second is the principle of symmetry.The laws of physics remain unchanged when observed from different coordinate systems. Therefore, the corresponding physical quantities and equations show equivalence under coordinate transformations. Maintaining equivalence not only improves data efficiency but also enhances generalization ability, which can significantly improve the performance of DeepH. The first-generation DeepH architecture simplified the equivalence problem through the local coordinate system and restored equivalent features through the transformation of local coordinates. The second-generation DeepH architecture is based on an equivalent neural network, named DeepH-E3. In this framework, the feature vectors of all input, hidden, and output layers are equivalent vectors. Recently, one of the authors of this work proposed DeepH-2, a new generation architecture for deep learning. DeepH-2 performs best in terms of efficiency and accuracy.
In summary, the deep learning model DeepH of this study was trained using the DeepH-2 method, containing a total of 17.28 million parameters. It formed a neural network that can be used for message passing based on 3 equivalent transformation blocks, and each node and edge carried 80 equivalent features.The embedding of the material structure includes atomic number and interatomic distance, and a Gaussian smoothing strategy is used, with the center range of the basis function ranging from 0.0 to 9.0 Å. The output features of the neural network are passed through a linear layer and then a DFT Hamiltonian is constructed through a Wigner-Eckart layer.
The study was trained on an NVIDIA A100 GPU for 343 epochs and 207 hours. Throughout the training process, the batch size was fixed to 1, which means that each batch contains one material structure. Finally, the initial learning rate was 4×10-4, the decay rate was 0.5, the decay patience was 20, the minimum selected learning rate was 1×10-5, and the training was stopped when the learning rate reached this value.
DeepH has excellent inference performance and can provide accurate band structure predictions
The mean absolute error (MAE) of the density functional theory Hamiltonian matrix elements predicted by the model reached 1.45, 2.35, and 2.20 meV on the training, validation, and test sets, respectively.This demonstrates the model’s ability to reason about unseen structures.
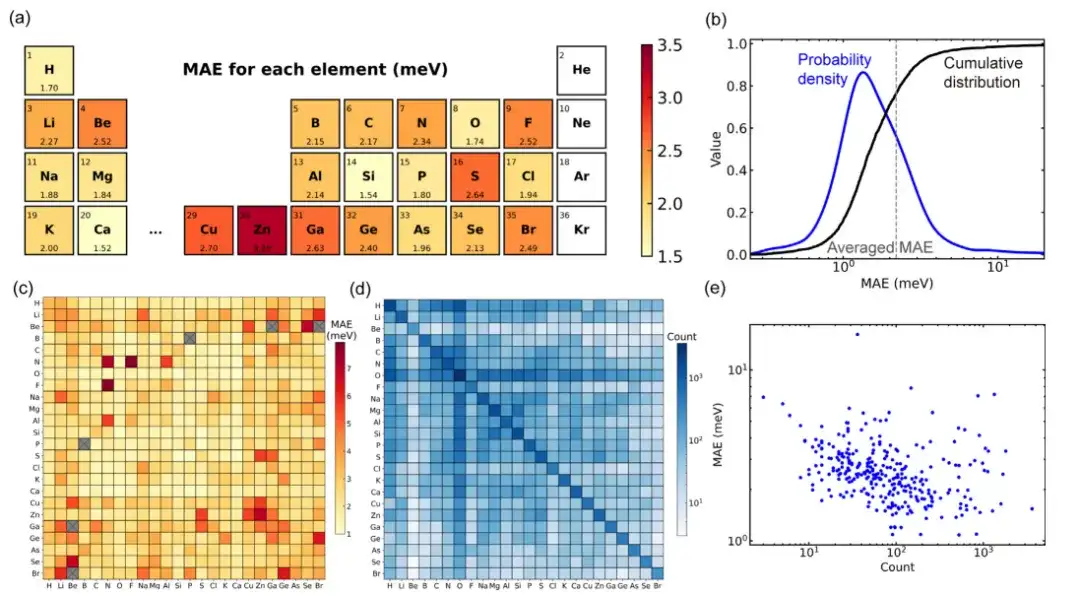
When evaluating the performance of the general material model trained by the Deep-2 method using a large material database of 104 solid materials, approximately 80% of all structures in the data set had a mean absolute error less than the average value (2.2 meV). Only 34 structures (about 1.4% of the test set) had a mean absolute error of more than 10 meV, indicating that the model has good prediction accuracy for mainstream structures.
By further analyzing the dataset, the deviation in the model’s performance on material structure may be due to the deviation in the dataset distribution.The study found that the more training structures of element pairs included in the dataset, the smaller the corresponding mean absolute error. This phenomenon may indicate that there is a "scaling law" for deep learning universal material models, that is, larger training datasets may improve model performance.
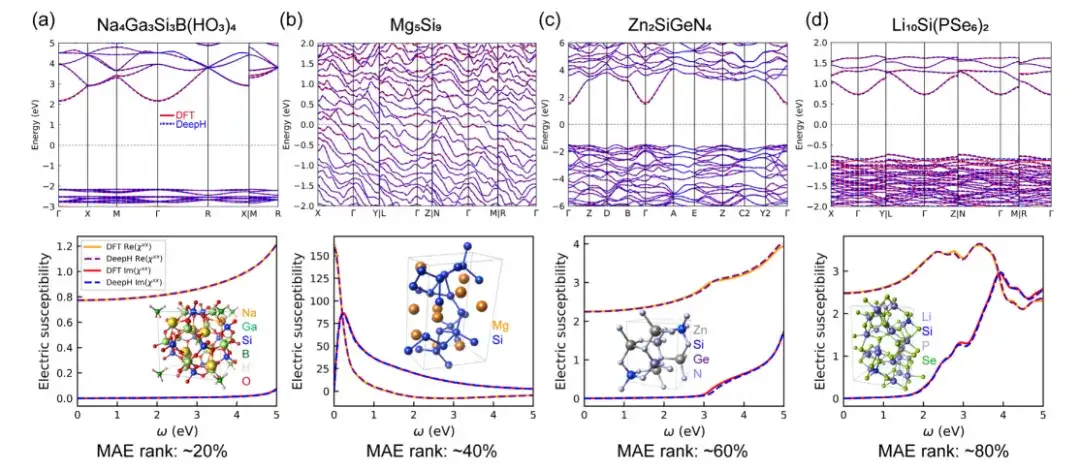
In order to evaluate the accuracy of DeepH universal material model in predicting material properties, the study used density functional theory (DFT) calculations and DeepH predicted DFT Hamiltonians in the calculation examples, and then compared the calculation results obtained by these two methods.The results predicted by DeepH are very close to those calculated by DFT, demonstrating the excellent predictive accuracy of DeepH in calculating material properties.
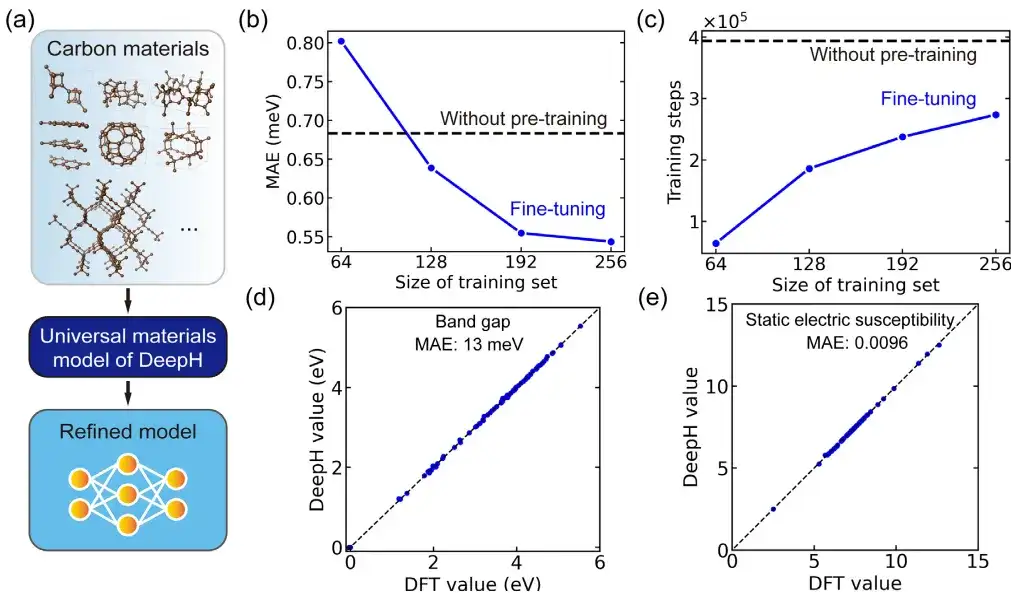
In the specific application, the study used a fine-tuned general material model to study carbon allotropes. The carbon material dataset comes from the Samara Carbon Allotrope Database (SACADA), which contains a total of 427 carbon allotropes with different atomic structures.
Based on this, the researchers fine-tuned the general material model and created an improved DeepH model specifically for carbon materials. Compared with the model without pre-training, fine-tuning can significantly reduce the mean absolute error of the predicted DFT Hamiltonian to 0.54 meV, and can also achieve comparable prediction accuracy in training structures with less than 50%.
In addition, fine-tuning also significantly improved training convergence and reduced training time. It can be said that fine-tuning helps improve prediction accuracy and enhance training efficiency. More importantly,The fine-tuned DeepH model showed significant advantages in predicting material properties. The fine-tuned model can provide accurate band structure predictions for almost all tested structures.
The material model is surging, AI4S has a long way to go
With ChatGPT as the starting point, AI has officially entered a new "big model era". This era is characterized by the use of huge data sets and advanced algorithms to train deep learning models that can handle complex tasks.In the field of materials science, these large models are combining with the wisdom of researchers to usher in an unprecedented new era of research.These large models can not only process and analyze massive amounts of scientific data, but also predict the properties and behavior of materials, thereby accelerating the discovery and development of new materials and driving this field towards a more efficient and precise direction.
Over the past period of time, AI for Science has been constantly colliding with materials science to create new sparks.
Based in China,The SF10 group of Beijing National Research Center for Condensed Matter Physics, the Institute of Physics of the Chinese Academy of Sciences, and the Computer Network Information Center of the Chinese Academy of Sciences worked together to feed tens of thousands of chemical synthesis path data to the large model LLAMA2-7b, thereby obtaining the MatChat model, which can be used to predict the synthesis path of inorganic materials; the University of Electronic Science and Technology of China, in collaboration with Fudan University and the Ningbo Institute of Materials Technology and Engineering of the Chinese Academy of Sciences, successfully developed "fatigue-resistant ferroelectric materials", taking the lead in the world to overcome the fatigue problem of ferroelectric materials that has plagued the field for more than 70 years; the AIMS-Lab of Shanghai Jiao Tong University has developed a new generation of intelligent material design model Alpha Mat. … Research results are emerging frequently, and material innovation and discovery have entered a new era.
Looking around the world,Google's DeepMind has developed GNoME, an artificial intelligence reinforcement learning model for materials science, which has found more than 380,000 thermodynamically stable crystalline materials, equivalent to "adding 800 years of intellectual accumulation to mankind", greatly accelerating the research speed of discovering new materials; Microsoft's MatterGen, an artificial intelligence generation model in the field of materials science, can predict the structure of new materials on demand based on the required material properties; Meta AI has cooperated with American universities to develop the industry's top catalytic material dataset Open Catalyst Project, and the organic metal framework adsorption dataset OpenDAC... Technology giants have stirred up the field of materials science with their own technologies.
Although AI has opened the door to exploring a wider range of material possibilities and significantly reduced the time and costs associated with material discovery compared to traditional material research and development methods, AI for Science still faces challenges in credibility and effective implementation in the field of materials. A series of issues remain to be addressed, including ensuring data quality, identifying and mitigating potential biases in data used to train AI systems, etc. This may also mean that there is still a long way to go before AI can play a greater role in the field of materials science.








