Command Palette
Search for a command to run...
Selected for ICML, Tsinghua AIR and Others Jointly Released the Protein Language Model ESM-AA, Surpassing the Traditional SOTA

As the driving force of countless biochemical reactions in cells, proteins play the role of architects and engineers in the microscopic world of cells. They not only catalyze life activities, but also serve as the basic building blocks for building and maintaining the morphology and function of organisms. It is the interaction and synergy between proteins that support the grand blueprint of life.
However, the structure of protein is complex and changeable, and traditional experimental methods are time-consuming and laborious to analyze protein structure. Protein language models (PLMs) came into being. By using deep learning technology, through analyzing a large amount of protein sequence data, learning the biochemical laws and co-evolution patterns of proteins, it has made remarkable achievements in the fields of protein structure prediction, adaptability prediction and protein design, and has greatly promoted the development of protein engineering.
Although PLMs have achieved great success at the residue scale, their ability to provide atomic-level information is limited. In response to this, Zhou Hao, an associate researcher at the Institute of Intelligent Industries at Tsinghua University, teamed up with Peking University, Nanjing University and Shuimu Molecular Team toA multi-scale protein language model ESM-AA (ESM All Atom) is proposed.By designing training mechanisms such as residue expansion and multi-scale position encoding, the ability to process atomic-scale information has been expanded.
ESM-AA has significantly improved the performance of tasks such as target-ligand binding, surpassing the current SOTA protein language models, such as ESM-2, and also surpassing the current SOTA molecular representation learning model Uni-Mol. Related research has been titled "ESM All-Atom: Multi-scale Protein Language Model for Unified Molecular Modeling".Published at ICML, the top machine learning conference.

Paper address:
https://icml.cc/virtual/2024/poster/35119
The open source project "awesome-ai4s" brings together more than 100 AI4S paper interpretations and provides massive data sets and tools:
https://github.com/hyperai/awesome-ai4s
Dataset: A mixed dataset of protein and molecular data was constructed
In the pre-training task,The study used a combined dataset of protein and molecular data containing structural information such as atomic coordinates.
For the protein dataset, the study used AlphaFold DB, which contains 8 million high-confidence AlphaFold2 predicted protein sequences and structures.
For the molecular dataset, the study used data generated by the ETKDG and MMFF molecular force fields, containing 19 million molecules and 209 million configurations.
When training ESM-AA, the researchers first mixed a protein dataset Dp and a molecule dataset Dm together as the final dataset, i.e., D=Dp∪Dm. For molecules from Dm, since they are composed only of atoms, their code conversion sequence X̄ is an ordered set of all atoms Ā without any residues, i.e., R̄=∅. It is worth noting that because molecular data is used in pre-training, ESM-AA can accept both proteins and molecules as input.
ESM-AA model construction: multi-scale pre-training and encoding to achieve unified molecular modeling
Inspired by the multi-language code switching method, ESM-AA first randomly decompresses some residues to generate multi-scale code switching protein sequences when performing prediction and protein design tasks. These sequences are then trained through carefully designed multi-scale positional encodings, and its effectiveness has been demonstrated at the residue and atomic scales.
When dealing with protein molecular tasks, that is, tasks involving proteins and small molecules, ESM-AA does not require any additional model assistance and can fully utilize the capabilities of the pre-trained model.
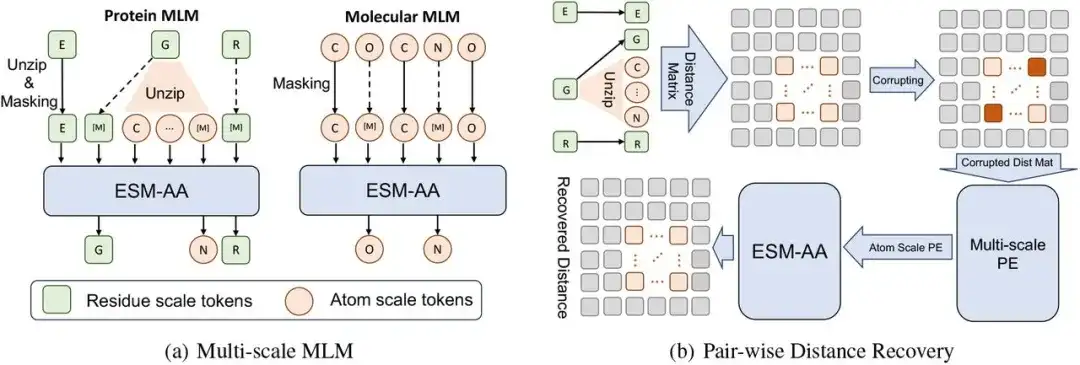
The multi-scale pre-training framework of this study consists of multi-scale masked language model (MLM) and pairwise distance recovery.
Specifically, at the residue scale, a protein X can be regarded as a sequence of L residues, that is, X = (r1,…,ri,…,rL). Each residue ri is composed of N atoms A Ai={a1i,…,aNi}. In order to construct the code switching protein sequence X̅, the study implemented a decompression process by randomly selecting a set of residues and inserting their corresponding atoms into X. In this process, the researchers arranged the decompressed atoms in order, and finally, after inserting the atom set Ai into X (i.e., decompressing residue ri), a code switching sequence X̄ was obtained.
Then,The researchers performed masked language modeling on the code switching sequence X̄.
First, a portion of atoms or residues in X̄ are randomly blocked, allowing the model to predict the original atoms or residues using the surrounding context. Then, the researchers used Paired Distance Recovery (PDR) as another pre-training task. That is, the atomic-scale structural information is destroyed by adding noise to the coordinates, and the destroyed interatomic distance information is used as the model input, requiring the model to restore the accurate Euclidean distance between these atoms.
Considering the semantic difference between the long-range structural information across different residues and the atomic-scale structural information within a single residue, this study only calculates the PDR within the residue, which can also enable ESM-AA to learn various structural knowledge within different residues.
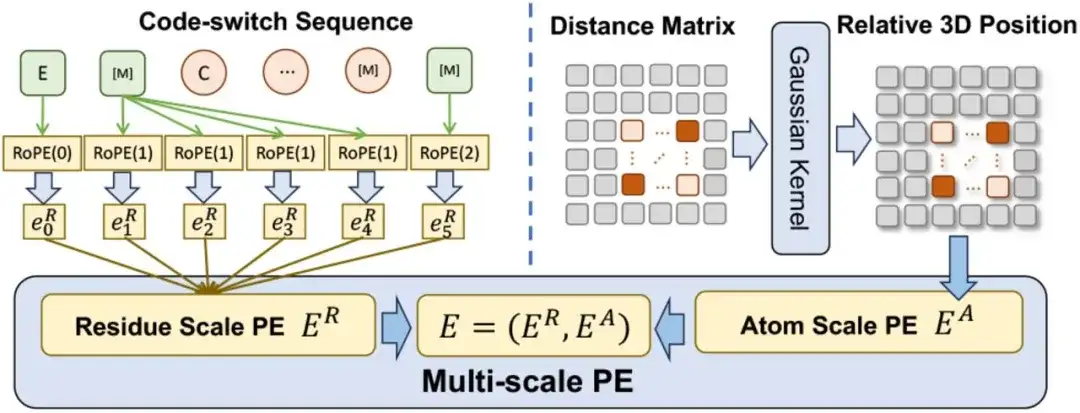
In terms of multi-scale position encoding, the researchers designed a multi-scale position encoding E to encode the position relationship in the code switching sequence. E contains a residue-scale position encoding ER and an atomic-scale position encoding EA.
For ER,The researchers extended an existing encoding method to enable it to encode residue-to-atom relationships while maintaining consistency with the original encoding when dealing with pure residue sequences.For EA,To capture the relationships between atoms, the study directly uses a spatial distance matrix to encode their three-dimensional positions.
It is worth mentioning that the multi-scale encoding method ensures that the pre-training will not be affected by the ambiguous position relationship, so that ESM-AA can work effectively at both scales.
When integrating multi-scale PE into Transformer, the study first replaced the sinusoidal coding in Transformer with the residual scale position coding ER, and regarded the atomic scale position coding EA as the bias term of the self-attention layer.
Research results: Integrating molecular knowledge to optimize protein understanding
To verify the effectiveness of the multi-scale unified pre-trained model, this study evaluated the performance of ESM-AA in various tasks involving proteins and small molecules.
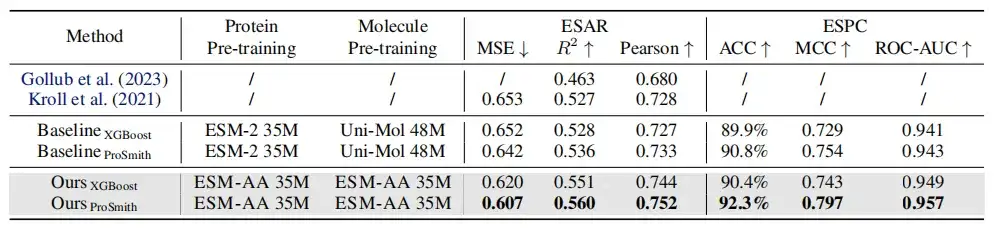
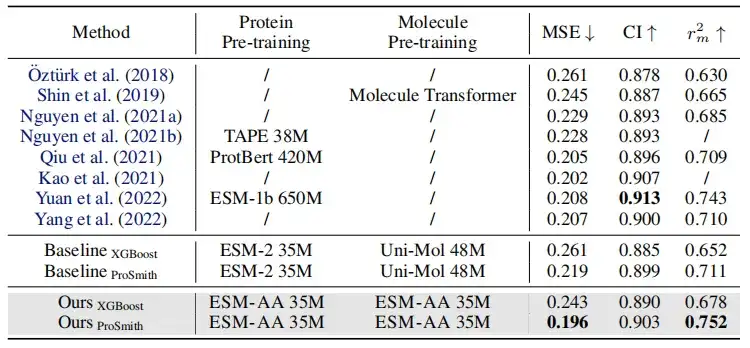
As shown in the table above, in the performance comparison of enzyme-substrate affinity regression task, enzyme-substrate pair classification task and drug-target affinity regression task,On most metrics, ESM-AA outperforms other models and achieves state-of-the-art results.Furthermore, fine-tuning strategies (such as ProSmith and XGBoost) built on ESM-AA consistently outperformed the versions combining two independent molecular pre-trained models with the protein pre-trained model (as shown in the last four rows of Tables 1 and 2 ).
It is worth noting thatESM-AA can even beat methods that use pre-trained models with larger parameter sizes.(For example, the comparison between the 5th, 7th and last rows in Table 2).
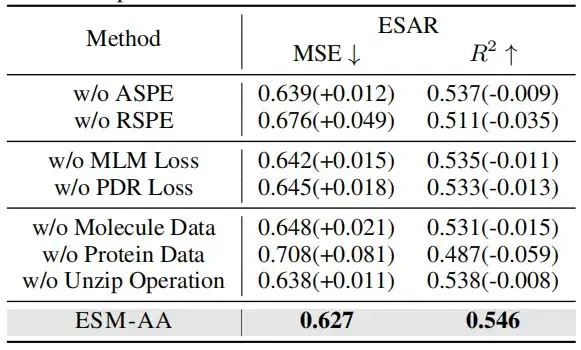
To verify the effectiveness of multi-scale position encoding, this study conducted ablation experiments in two cases: one is without using atomic scale position encoding (ASPE); the other is without using genetic scale position encoding (RSPE).
When either the molecule or protein data were removed, the model performance dropped significantly. Interestingly, the performance drop was more pronounced when the protein data was removed than when the molecule data was removed. This suggests that when the model is not trained on protein data, it quickly loses protein-related knowledge, resulting in a significant drop in overall performance. However,Even without molecular data, the model can still obtain atomic-level information through decompression operations.
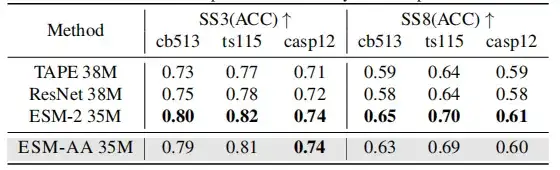
Since ESM-AA was developed based on existing PLMs, this study hopes to determine whether it still retains a comprehensive understanding of proteins, thereby testing the ability of protein pre-trained models in protein structure understanding by using secondary structure prediction and unsupervised contact prediction tasks.
The results show that although ESM-AA may not achieve optimal performance in this type of study,However, its performance in secondary structure prediction and contact prediction is similar to that of ESM-2.
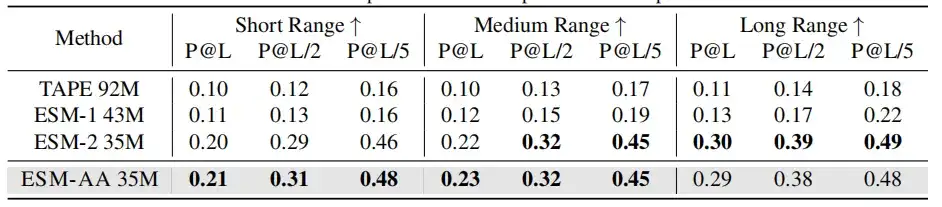
In molecular benchmarking,ESM-AA performs comparably to Uni-Mol in most tasks.It outperforms several molecule-specific models in many cases, demonstrating its potential as a powerful approach for molecular tasks.
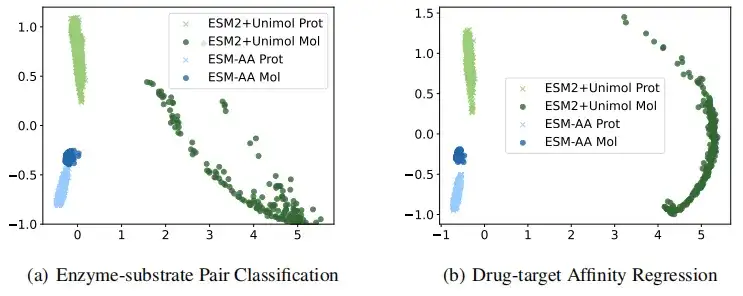
In order to more intuitively illustrate that ESM-AA obtains higher quality protein and small molecule representations, this study visually compared the representations extracted by ESM-AA and ESM-2+Uni-Mol in the tasks of enzyme-substrate pair classification and drug target affinity regression. The results showed thatThe ESM-AA model is able to create more cohesive semantic representations that encompass both protein and molecular data, which makes ESM-AA outperform the two separate pre-trained models.
Protein language model, the next step of the big language model
Since the 1970s, more and more scientists have believed that "the 21st century is the century of biology." Last July, Forbes wrote a long article that LLM puts people at the forefront of a new round of changes in the field of biology. Biology turns out to be a system that can be deciphered, programmable, and even digital in some aspects.LLM, with its amazing ability to control natural languages, provides the potential for deciphering biological languages.This also makes protein language model one of the most popular areas of this era.
The protein language model represents the cutting-edge application of AI technology in biology. By learning the patterns and structures of protein sequences, it can predict the function and morphology of proteins, which is of great significance for new drug development, disease treatment and basic biological research.
Previously, protein language models such as ESM-2 and ESMFold have demonstrated comparable accuracy to AlphaFold, with faster processing speeds and more accurate prediction capabilities for "orphan proteins." This not only speeds up the prediction of protein structure, but also provides new tools for protein engineering, allowing researchers to design new protein sequences with specific functions.
Furthermore, the development of protein language models benefits from the so-called “scaling laws”.That is, the performance of the model improves significantly with the increase of model scale, dataset size and computational effort.This means that with the increase of model parameters and the accumulation of training data, the capabilities of the protein language model will achieve a qualitative leap.
In the past two years, protein language models have also entered a period of rapid development in the business community. In July 2023, Baidu Bio and Tsinghua University jointly proposed a model called xTrimo Protein General Language Model (xTrimoPGLM), which has a parameter volume of up to 100 billion (100B) and significantly outperforms other advanced baseline models in a variety of protein understanding tasks (13 out of 15 tasks).On the generation task, xTrimoPGLM is able to generate new protein sequences that are similar to natural protein structures.
In June 2024, AI protein company Tushen Zhihe announced thatThe first natural language protein model in China, TourSynbio™, was made open source to all researchers and developers.This model achieves an understanding of protein literature in a conversational manner, including functions such as protein properties, function prediction and protein design. In terms of evaluation indicators of comparative protein evaluation datasets, it surpasses GPT4 and becomes the first in the industry.
In addition, the breakthrough in technology research represented by ESM-AA may also mean that the development of technology is about to pass the "Wright Brothers moment" and usher in a leap. At the same time, the application of protein language models will not only be limited to the medical and biopharmaceutical fields, but may also be extended to agriculture, industry, materials science, environmental remediation and other fields, promoting technological innovation in these fields and bringing unprecedented changes to mankind.








