Command Palette
Search for a command to run...
A Cell Model With 100 Million Parameters Is Here! In a Nature Journal, a Tsinghua University Team Released scFoundation: Simultaneous Modeling of 20,000 Genes
In recent years, large-scale pre-trained models are leading a new wave of artificial intelligence. "Big models" can serve diverse tasks in different fields as "basic models" by extracting deep rules from large-scale, multi-source data. For example, large language models have mastered the ability to understand and recognize language by learning from large amounts of text data, revolutionizing the paradigm in the field of natural language processing.
Similarly, in the field of life sciences, organisms also have their own "underlying language" - cells are the basic structural and functional units of the human body.If DNA, RNA, protein and gene expression values are compared to "words", they are combined together to form the sentence "cell".Therefore, if we can develop an artificial intelligence cell model based on the cell "language", it will hopefully provide a new research paradigm and revolutionary research tools for life sciences and medicine.
However,There are currently three main challenges in training large-scale single-cell data:
* Gene expression pre-training data needs to cover cell landscapes of different states and types. Currently, most single-cell RNA sequencing (scRNA-seq) data are loosely organized, and a comprehensive and complete database is still missing;
* During training, traditional Transformers have difficulty processing “sentences” consisting of nearly 20,000 protein-coding genes;
* scRNA-seq data from different technologies and laboratories vary in sequencing depth, which prevents the model from learning unified and meaningful cell and gene representations.
To address these challenges,Professor Zhang Xuegong, Director of the Life Basic Model Laboratory of the Department of Automation, Tsinghua University, Professor Ma Jianzhu of the Department of Electronics/AIR, and Dr. Song Le of Bio-Technology collaborated on the research.In June 2024, a research paper titled "Large-scale foundation model on single-cell transcriptomics" was published in Nature Methods.
The paper introduces a large cell model called scFoundation, which can process about 20,000 genes simultaneously.As a basic model, it demonstrates outstanding performance improvements in a variety of biomedical downstream tasks such as cell sequencing depth enhancement, cell drug response prediction, and cell perturbation prediction, providing a new paradigm for artificial intelligence in single-cell research.
Research highlights:
The scFoundation cell model is trained based on gene expression data from 50 million cells, has 100 million parameters, and can process approximately 20,000 genes simultaneously* The model uses an asymmetric design to reduce computational and memory challenges* The model provides new research ideas for gene network inference and transcription factor identification

Paper address:
https://www.nature.com/articles/s41592-024-02305-7
The open source project "awesome-ai4s" brings together more than 100 AI4S paper interpretations and also provides massive data sets and tools:
https://github.com/hyperai/awesome-ai4s
Datasets: Building a comprehensive single-cell dataset
The researchers built a comprehensive single-cell dataset by collecting all publicly available single-cell resource data.These include Gene Expression Omnibus (GEO), Single Cell Portal, HCA, Human Genome Project (hECA), Deeply Integrated Human Single-Cell Omics Data (DISCO), European Molecular Biology Laboratory-European Bioinformatics Institute Database (EMBL-EBI), etc.
* GEO download address:https://www.ncbi.nlm.nih.gov/geo/
* Single Cell Portal Download Link:https://singlecell.broadinstitute.org/single_cell
* HCA download address:https://data.humancellatlas.org/
* EMBL-EBI download address:https://www.ebi.ac.uk/
The researchers aligned all data with the gene list of 19,264 protein-coding and common mitochondrial genes determined by the HUGO Gene Nomenclature Committee.More than 50 million human scRNA-seq data were obtained for pre-training.
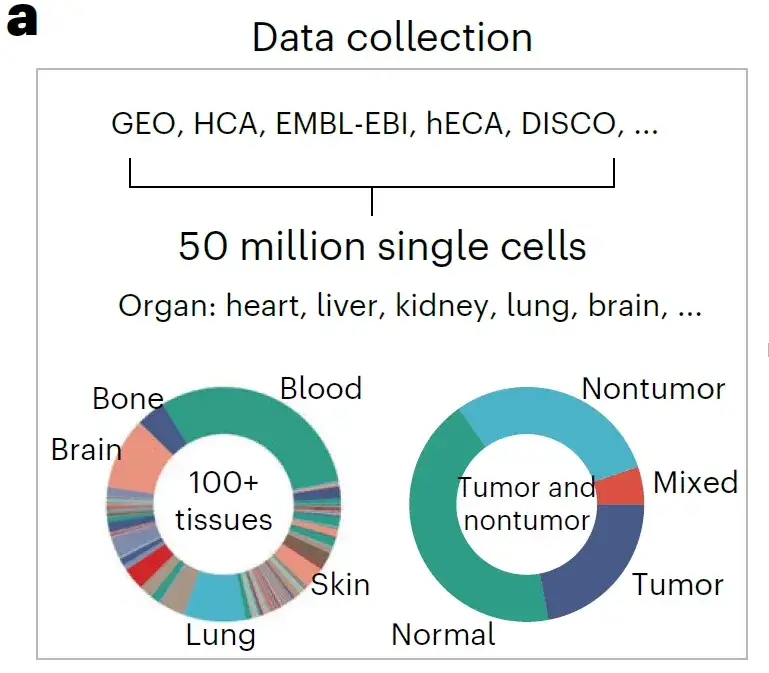
A rich source of data builds a pre-trained dataset rich in biological patterns. Anatomically, it spans more than 100 tissue types, covering a variety of diseases, tumors, and normal states, including almost all known human cell types and states, as shown in the figure above.
Model architecture: Building a 100 million parameter scFoundation model
The scFoundation model developed by the researchers has approximately 100 million parameters, and its parameter scale, gene coverage, and data scale are "among the best" in the single-cell field.
Model design
The researchers developed the xTrimoGene model as the backbone model of scFoundation, which is a scalable Transformer-based model that includes an embedding module and an asymmetric encoder-decoder structure.
Among them, the vector module converts continuous gene expression scalar values into learnable high-dimensional vectors, ensuring that the original expression values are fully preserved; the encoder takes nonzero and nonmasked expression genes as input, uses a vanilla transformer block and has a large number of parameters; the decoder takes all genes as input, uses a performer block and has a relatively small number of parameters.
This asymmetric design reduces computational and memory challenges compared to other architectures.Data shows that the module only requires 3.4% of the traditional language model Transformer while maintaining the same parameter scale.
Pre-training tasks
The researchers designed a pre-training task called RDA (read-depth-aware) modeling.This is an extension of the masked language model that takes into account the high variance of sequencing depth in large-scale data.
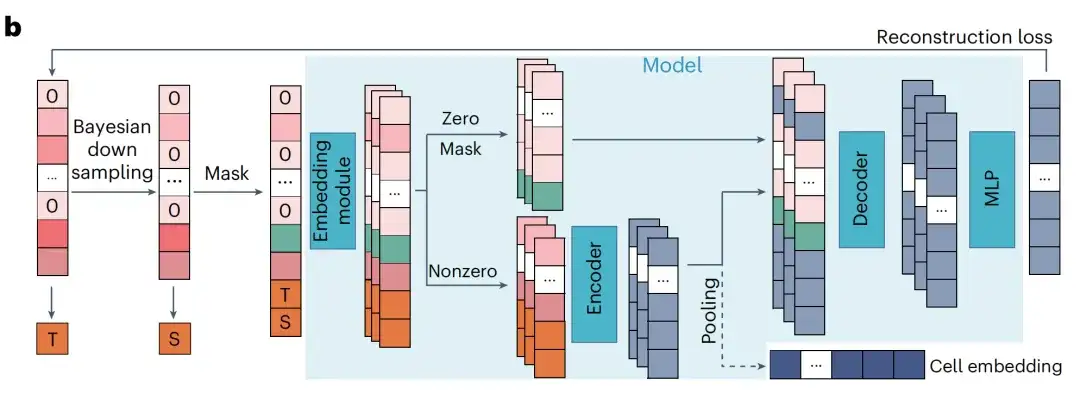
In RDA modeling, the model predicts the expression of masked genes based on the context genes of the cell. The researchers regarded the total count as the sequencing depth of a cell and defined two total count indicators: T (target) and S (source), which represent the total counts of the original sample and the input sample, respectively. The researchers randomly masked the zero-expression and non-zero-expression genes in the input sample and recorded their indexes.
The model then uses the masked input sample and two metrics to predict the expression value of the original sample at the masked index. This enables the pre-trained model to not only capture gene relationships within cells, but also coordinate cells of different sequencing depths. During inference, the researchers input the raw gene expression of the cell into the pre-trained model and set T higher than its total count S to generate gene expression values with enhanced sequencing depth.
Simply put, RDA can downsample the sequencing depth, so that in addition to completing the traditional mask recovery task, the model can also recover the gene expression information of high-quality cells from low-quality cells during the pre-training stage.
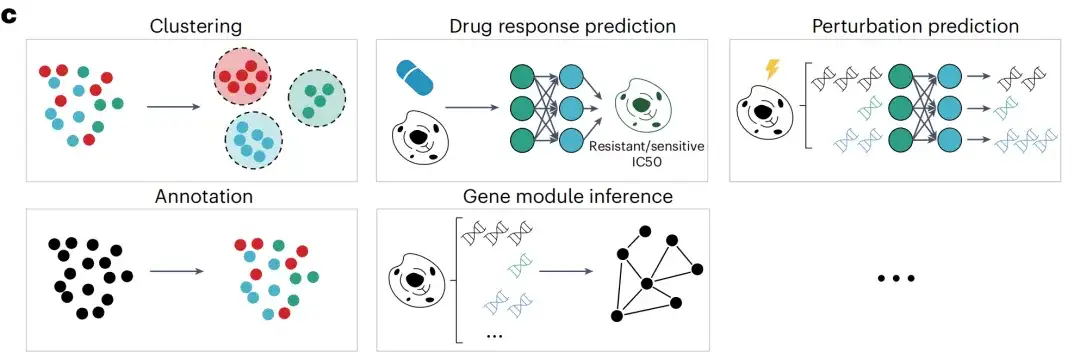
After pre-training, the researchers further applied the scFoundation model to multiple downstream tasks. The output of the scFoundation encoder is summarized as a cell-level vector for cell-level tasks, including clustering (within and across datasets), batch and single-cell drug response prediction, and cell type annotation. The output of the scFoundation decoder is a gene-level context vector for gene-level tasks such as perturbation prediction and gene module inference.
Research results: scFoundation models have superior performance
In actual applications, the scFoundation model supports two modes: "out-of-the-box" and "fine-tuning".In "out-of-the-box" mode, thanks to its unique pre-trained tasks, the model can be directly used to improve the quality of cell data, and can reach or exceed the results of existing methods without further adjustment. In addition, users can use scFoundation to extract pre-trained representations of cells, which can be used to identify cell type-specific gene modules and transcription factors, and can be widely used in downstream tasks.
Scalable, fine-tuning-free sequencing deep enhancement model
The researchers trained three models with 3M, 10M, and 100M parameters respectively, and recorded their losses on the validation dataset.
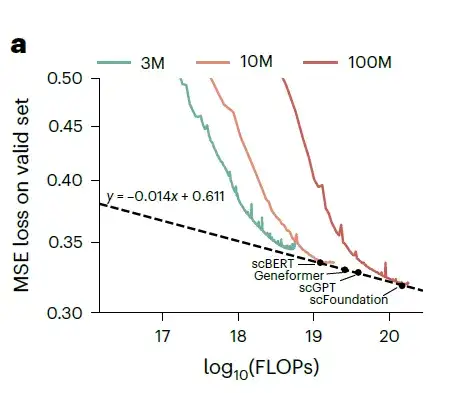
Training loss under different parameter sizes and FLOPs. The green curve represents the 3M model, the orange curve represents the 10M model, and the red curve represents the 100M model.
As the model parameters and floating point operations (FLOPs) increase, the loss on the validation dataset shows a power law decrease. The researchers then estimated the performance of xTrimoGene architecture models of various sizes and compared them with scVI. As shown in the figure above,The scFoundation model with 100 million parameters performed best among all the models.The researchers further evaluated the three models on the cell type annotation task and observed that performance improved as the model size increased.
The researchers evaluated this capability on independent test data of 10,000 cells randomly selected from the validation dataset, which downsampled the total counts to 1%, 5%, 10%, and 20% of the original data, generating four datasets with different total count changes. For each dataset, the mean absolute error (MAE), mean relative error (MRE), and Pearson correlation coefficient (PCC) between the predicted value and the actual non-zero gene expression were measured using untuned scFoundation.
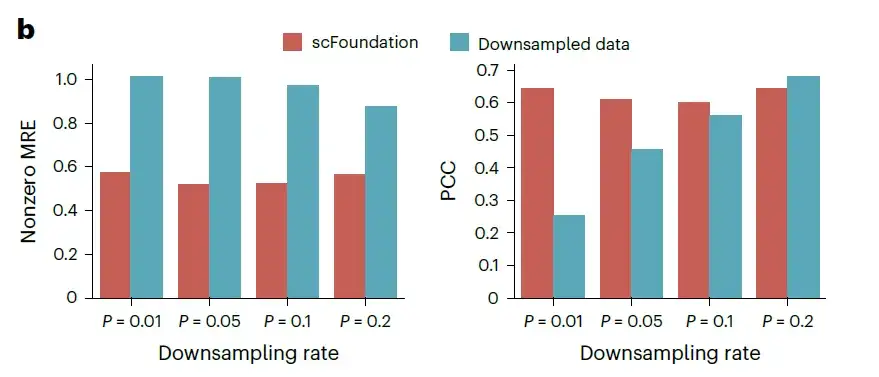
Evaluation of read depth enhancement performance on unknown datasets MRE and PCC were used to evaluate the performance of recovered gene expression, with lower MRE and higher PCC indicating better performance.
As shown in the figure above, even when the downsampling rate is lower than 10%, the MAE and MRE of scFoundation are significantly reduced by half.These results demonstrate the ability of scFoundation to enhance gene expression at extremely low total counts.
Downstream tasks - cancer drug response prediction tasks
Cancer drug responses (CDRs) aim to study the response of tumor cells to drug intervention. Computational prediction of CDRs is crucial for guiding anticancer drug design and understanding cancer biology. In this study, researchers combined scFoundation with the CDR prediction method DeepCDR to predict the half-maximal inhibitory concentration IC50 value of drugs in multiple cell line data to verify whether scFoundation can provide useful embedding information for overall gene expression data based on single-cell training.
The researchers evaluated the performance of scFoundation-based results versus gene expression-based results across multiple drugs and cancer cell lines.The results showed that most drugs and all cancer types achieved higher Pearson correlation coefficients (PCC) using scFoundation embedding.As shown in the following figure:
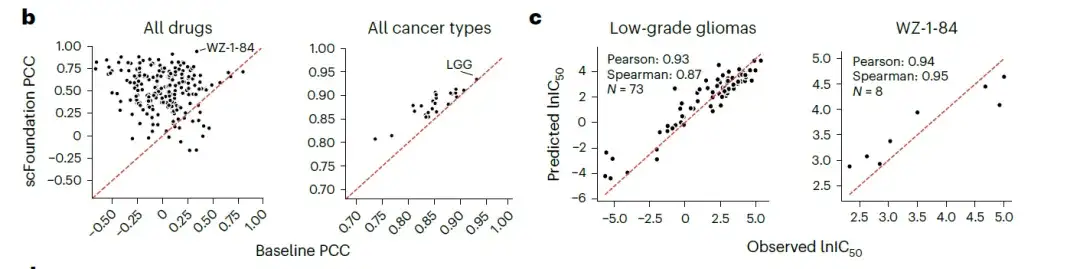
- Note: The Pearson correlation coefficient is a statistic that measures the strength of the linear relationship between variables, and its value range is between -1 and 1. A correlation coefficient close to 1 indicates that there is a completely positive linear relationship between the two variables; close to -1, it indicates that there is a completely negative linear relationship; close to 0, it indicates that there is no linear relationship between the two variables.
This shows thatAlthough scFoundation is pre-trained on single-cell transcriptome data, the learned gene relationships can be transferred to global expression data.Generates compressed vectors, facilitating more accurate IC50 predictions. scFoundation thus has the potential to expand our understanding of drug responses in cancer biology and guide the design of more effective anticancer treatments.
Downstream task - single cell drug response classification task
Inferring drug sensitivity at the single-cell level helps identify specific cell subtypes that exhibit different drug resistance profiles, providing valuable insights into underlying mechanisms and new therapies. Therefore, the researchers applied scFoundation to the critical task of single-cell drug response classification, based on a downstream model called SCAD.
The researchers focused on four drugs (sorafenib, NVP-TAE684, PLX4720, and etoposide) that showed low AUC (Area Under Curve) values in the original study. The scFoundation-based model was compared with the baseline SCAD model that used all gene expression values as input. The results showed that the scFoundation-based model achieved higher scores in the AUC values of all drugs, especially for NVP-TAE684 and sorafenib, with AUC values increased by more than 0.2, as shown in the figure below.
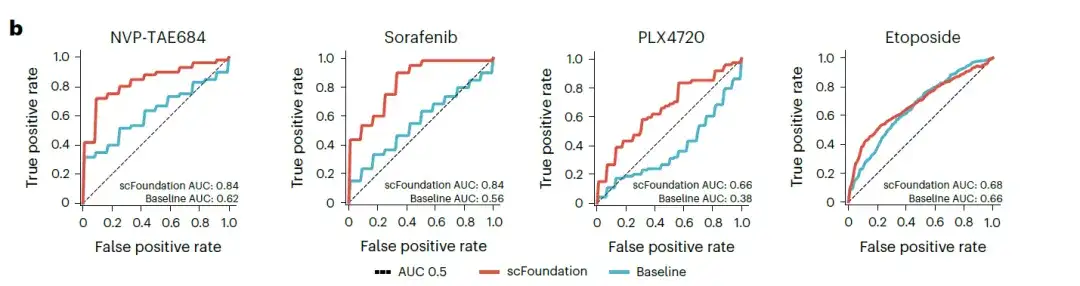
Note: AUC can be used to measure the performance of the model. The value range of AUC is 0 to 1. The larger the value, the better the model classification performance.
These results validate the potential of using scFoundation embedding to capture drug sensitivity biomarker signals.
Downstream task - cell perturbation prediction task
Understanding the response of cells to perturbations is crucial for biomedical applications and drug design, helping to identify gene-gene interactions and potential drug targets between different cell types. The researchers combined scFoundation with an advanced model, GEARS, to predict perturbation responses at single-cell resolution, and calculated the mean mean square error (MSE) of the top 20 differentially expressed (DE) genes with significant differences in gene expression profiles before and after as an evaluation criterion.
The results show thatCompared with the original GEARS baseline model, the scFoundation-based model achieved lower MSE values.The figure below shows the expression changes of the top 20 genes in the double gene perturbation ETS2 + CEBPE:
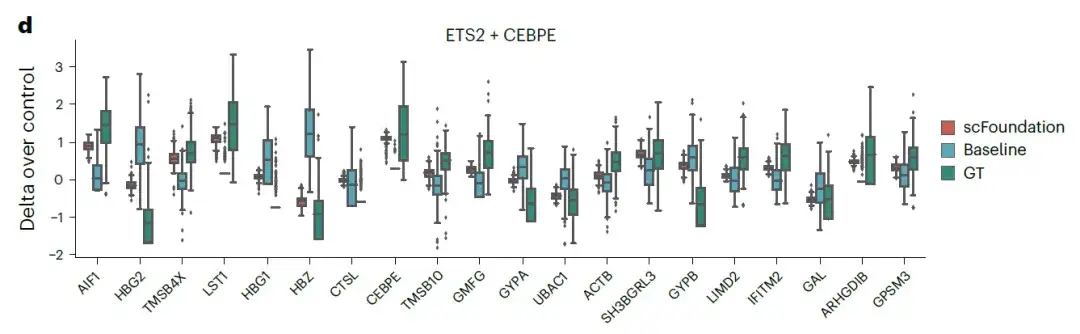
These results suggest that by extracting gene representations from single cells to construct specific gene co-expression networks,scFoundation successfully captured the cellular and gene characteristics under different conditions and significantly improved the accuracy of single/double perturbation predictions.
In summary, the scFoundation model provides new ideas and methods for establishing the model architecture, training framework, and downstream demonstration application system of large cell pre-training models, provides basic functions for learning biomedical tasks, and expands the boundaries of basic models in the single-cell field.
Exploring life science big models with better performance
Demis Hassabis, CEO and founder of DeepMind, a world-leading artificial intelligence company, once said:“At the most basic level, biology can be viewed as a very complex and dynamic information processing system. Just as mathematics proved to be the right descriptive language for physics, biology may be a perfect domain for artificial intelligence applications.”
However, traditional AI methods require a large amount of labeled data to make accurate predictions. But in the field of life sciences, high-quality labeled data is often in short supply. To build more accurate downstream task models based on less data means that the underlying basic models need to have better representation or general capabilities. Therefore, more and more researchers are beginning to work on designing better vertical large models in the biological field.
May 2023A research team from the University of Toronto released the first large-scale language model based on single-cell biology, scGPT.It is pre-trained on more than 10 million cells, and the model can achieve transfer learning across various downstream tasks. In July of the same year, the team further attempted to update scGPT by generating pre-training on more than 33 million cells. The results showed that scGPT can effectively extract key biological insights about genes and cells and achieve advanced performance in various downstream tasks, including multi-batch integration, multi-omics integration, cell type annotation, genetic perturbation prediction, and gene network inference.
The study, titled “scGPT: toward building a foundation model for single-cell multi-omics using generative AI,” was published in Nature Methods.
* Paper link:https://www.nature.com/articles/s41592-024-02201-0
September 2023The Xcompass Consortium, a multidisciplinary research team from the Chinese Academy of Sciences, has successfully constructed the world's first cross-species life foundation model - GeneCompass.The model integrates transcriptome data of more than 126 million single cells from humans and mice, fuses four types of prior knowledge including promoter sequences and gene co-expression relationships, and has 130 million basic model parameters, achieving a panoramic learning and understanding of the laws of gene expression regulation, while supporting the prediction of cell state changes and accurate analysis of various life processes.

The study was published on bioRxiv under the title "GeneCompass: Deciphering Universal Gene Regulatory Mechanisms with Knowledge-Informed Cross-Species Foundation Model."
In October 2023, global pharmaceutical giant Sanofi announced a large-scale strategic partnership with BioMap BioScience. The two parties will jointly develop cutting-edge models for biotherapeutic drug discovery based on BioMap's Life Science AI Foundation Model.
Looking into the future, applying the complex understanding and innovative generation capabilities of large language models that far exceed human imagination to the more complex "natural language" of life will hopefully truly change the research paradigm of life sciences.
References:
1.https://www.jiqizhixin.com/articles/2023-9-29
2.https://www.tsinghua.edu.cn/info/1175/112118.htm
3.https://hope.huanqiu.com/article/4FYZxnpu88J
4.https://www.jiqizhixin.com/articles/2023-7-5-26'








