Command Palette
Search for a command to run...
Li Huashan and Wang Biao's Research Group at Sun Yat-sen University Developed the SEN Machine Learning Model to Predict Material Properties With High Accuracy

Contents at a Glance:Understanding global crystal symmetry and analyzing equivariant information are crucial for predicting material properties, but existing convolutional network-based algorithms cannot fully meet these requirements. In response to this, Li Huashan and Wang Biao's research group at Sun Yat-sen University developed a machine learning model called SEN, which accurately perceives the interaction between intrinsic crystal symmetry and material structure clusters.
Keywords: Material properties prediction deep learning MP database
Author | Li Baozhu
Editor | Sanyang
Crystal symmetry plays a key role in studying the physical properties of materials, understanding crystal structures, designing new materials, and conducting experiments such as X-ray diffraction. Understanding crystal symmetry helps simplify analysis, better understand material properties, and improve the efficiency of calculating material performance. More importantly, crystal symmetry can also directly affect the material's charge distribution, optical properties, magnetic properties and other physical properties.
In recent years, machine learning based on statistical mechanisms has been widely used. From the perspective of machine learning, crystal symmetry can be regarded as the invariance and equivariance of materials. However, the existing machine learning algorithms for crystal materials based on advanced graph networks find it difficult to encode complex material invariance and equivariance.
In addition, although the Stacked Capsule Autoencoder (SCAE) can also directly extract spatial symmetry features from the original data, the traditional capsule model is still unable to analyze the relationship between the structure and performance of complex material systems.
In view of the above challenges,The research group led by Huashan Li and Biao Wang from Sun Yat-sen University developed a machine learning model called SEN (symmetry-enhanced equivariance network)., overcoming the poor performance of convolution-based algorithms in high-symmetry space groups, and achieving high-precision material property predictions in all space groups. Currently, the relevant results have been published in "Nature Communications".

Related results have been published in "Nature Communications"
Get the paper:
https://www.nature.com/articles/s41467-023-40756-2
01 Dataset: 6,027 crystal materials in the MP database
The researchers extracted the characteristics of crystalline materials based on the concept of chemical environment and the representation method of graphical models. They defined their chemical environment by the surrounding atoms and bonds within the cutoff radius of the target atom, and extracted the atom type, atomic connectivity and bond length around each atom from the Materials Project, an open source Python database for materials analysis.
It is reported that,The data sets used to predict band gap and formation energy in this study are from the Materials Project database, and the data sets of band gap and formation energy contain 6,027 (divided into training set, validation set, and test set in a ratio of 8:1:1) and 30,000 materials, respectively.The two data sets consist of 64 elements, covering the elements in the periodic table except the noble gas group, lanthanides, actinides and radioactive elements.
The researchers used density functional theory (DFT) calculations to predict the composition of 6,027 crystalline materials in the Materials Project database and tested the performance of the SEN model based on the predicted conclusions.
The crystal symmetry and chemical environment data used in this study are available from the Zenodo database.
Visit the link:
https://doi.org/10.5281/zenodo.8142678
02 Model architecture: unified training of 3 modules
As shown in the figure below,The SEN model adopts a complex deep learning architecture, which includes feature extraction (FE), symmetry perception (SP) and property prediction (PP) modules.
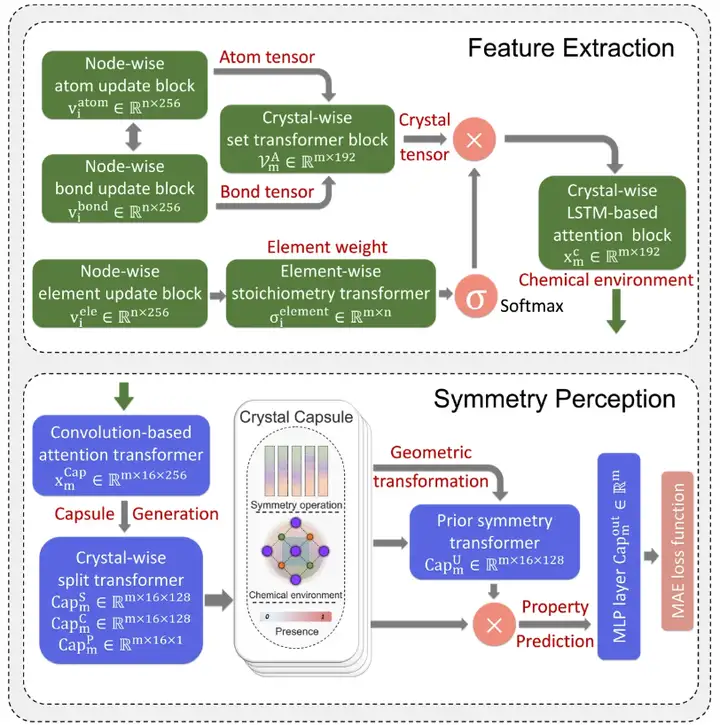
The SEN architecture consists of feature extraction, symmetry perception, and attribute prediction modules.
In this study, the research team achieved accurate prediction of the properties of multiple materials through unified training of three modules, and described the interaction between atoms through the SEN model.
First, the feature extraction module senses the input atomic and chemical bond data, which includes the information of N atoms and M bonds in the original unit of the target material. Finally, through the high-throughput screening process, a material dataset including stoichiometry, crystal structure, atomic information and bond information is constructed.
Using the material dataset as the only input data for the SEN model, the researchers simultaneously calculated the atomic chemical environment vector VmA and the element weight vector VmE based on the structural data and stoichiometric data.
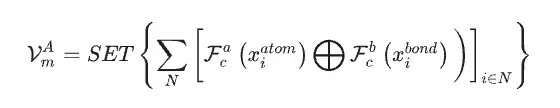
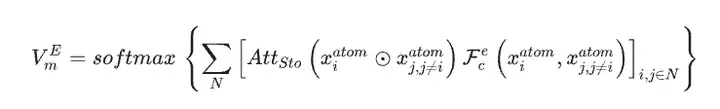
After activation by the multi-layer perceptron, the element weight vector is converted into the probability vector of the corresponding atom. The researchers then updated all atomic-level correlations through element-wise operations between the atomic chemical environment vector and the element weight vector, thereby obtaining the chemical environment matrix of the material through the LSTM-attention layer.
Secondly, the study innovatively applied the capsule mechanism to the prediction of material properties. Through the symmetry perception module designed based on the capsule mechanism, the material chemical environment is converted into a material capsule consisting of a symmetry operator, a convolution material chemical environment, and an existence value to perceive and preserve the crystal symmetry. Furthermore, by performing symmetry operations on the material's chemical environment matrix, different symmetry patterns can be generalized to the crystal capsule.
Finally, in terms of property prediction, the SEN model predicts the target material properties through an MLP-based mapping function.
03 SEN model predicts material properties with high accuracy
Conclusion 1: The SEN model accurately perceives atomic interaction information
To verify the effectiveness of the feature extraction module, the researchers trained SEN's ability to predict the band gap of crystalline materials until the mean absolute error (MAE) was less than 0.15 eV, and then analyzed the chemical environment intermediate data generated by the feature extraction module.
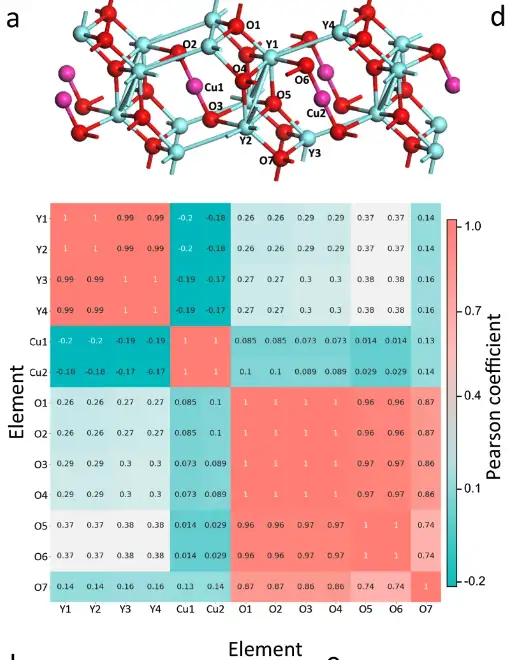
Atom-based chemical environment correlation analysis
Specifically, the researchers extracted the chemical environment matrix of each atom in the primitive cell of Y4Cu2O7. The Pearson coefficient between the atomic matrices was calculated, generating the correlation analysis graph shown above. The Pearson coefficient between atoms in the same element group is much larger than that between atoms in different element groups, so the three element groups in Y4Cu2O7 can be clearly distinguished.
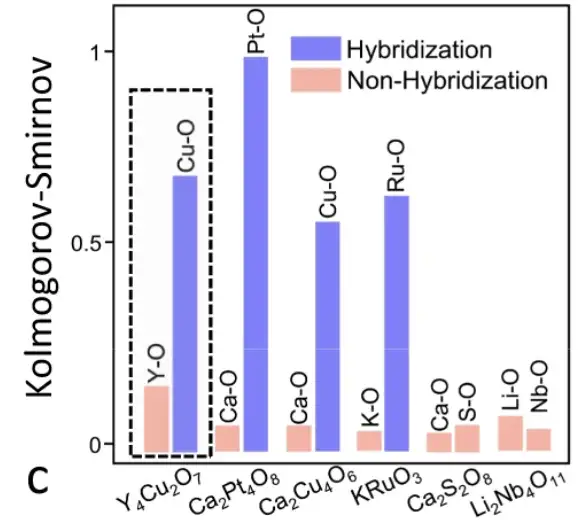
The atomic correlations of six materials were learned by the SEN model
As shown in the figure above, the SEN model has learned and encoded atomic interaction information and successfully detected the hybridization phenomenon, which is of great significance for the prediction of electronic properties.
Conclusion 2: The prediction performance of the SEN model is better than that of MegNet
To study the mapping from chemical environment to material properties in the SEN model, the researchers selected five materials from the MP database - Be(6)Ni(2), Sr(4)Ge(2)S(8), Li(2)V(2)F(12), CsAsF(6), and BaB(2)F(8), with band gaps of 0 eV, 3.25 eV, 4.86 eV, 7.24 eV, and 10.12 eV, respectively.
It is observed that there is a strong correlation between the band gap and the PDF (probability density function) of the material chemical environment, that is, as the band gap increases, the PDF gradually spreads. The projection of the entire data set from the material chemical environment to the band gap is shown in the figure below. The 6,027 crystal materials are evenly distributed in the main feature space, and the change of the band gap is continuous and monotonic in the entire space.
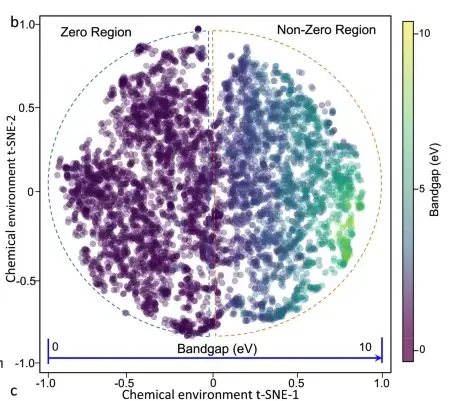
2D t-SNE plot of 6027 materials. The color of the circle indicates the band gap value.
To verify that the feature-attribute relationships learned by the machine learning model are consistent with basic physical principles, the researchers generated a 2D t-SNE map of the chemical environment of the Ca-OX material and investigated various material characteristics (composition, point group, spin polarization, etc.). They finally found that the material band gap depends on complex material characteristics and cannot be simply predicted by any key factor.
Nevertheless, the SEN model achieves significant improvements in bandgap prediction.The SEN model achieves a mean square error (MAE) of 0.25 eV when predicting the band gap of materials in the test dataset, which is a significant improvement over the MAE obtained by models with MLP, DenseNet, TFN, SE(3), and EGNN modules on the test dataset.
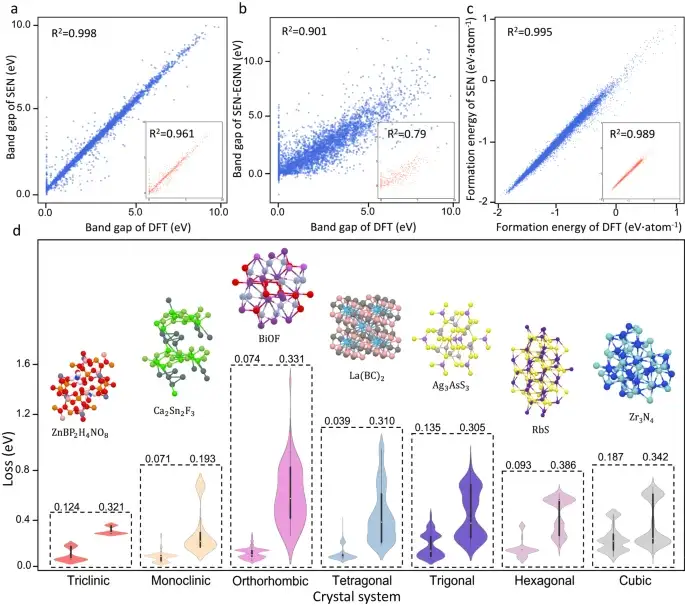
Prediction of properties of crystal materials with different symmetry
As shown in Figure d above, the researchers compared the prediction quality of the SEN model and the MegNet21 model (general material network model) for different crystal systems, further revealing the significant impact of symmetry perception on the prediction of material properties.From the error distribution plots, the prediction performance of the SEN model is better than that of MegNet in all crystal systems.
In addition, the SEN model significantly reduces the effective feature dimension by being aware of the full crystal symmetry. This feature elimination process alleviates the overfitting problem and strengthens the mapping from material features to properties.
The paper shows thatThe mean absolute errors of the band gap and formation energy predicted by the SEN model are approximately 22.9% and 38.3% lower than those of common machine learning models, respectively.
04 AI promotes the transformation and development of the materials industry
For a long time, the design, research and development of new materials and the reform of material performance have been one of the driving forces for scientific and technological progress, playing an important role in many fields such as electronics, energy, medical care, aerospace, etc. However, the traditional material research and development process often requires a large number of experiments to continuously correct performance and improve feasibility. This process is long and requires a huge amount of manpower and financial resources.
With the accelerated application of AI, AI for Science has received more and more attention, and its combination with materials has become a new direction for more and more scholars and companies to explore. On the one hand, AI can analyze large amounts of data and perform simulation predictions, thereby accelerating the discovery of new materials and optimizing their performance; on the other hand, materials science has also become an important foothold for key AI technologies such as machine learning, natural language processing, and high-performance computing.
It can be said that AI is quietly changing the design and application of new materials. In the future, with the continuous iteration of more powerful AI models and the update and expansion of material databases under data sharing, AI is bound to further promote the birth of new materials.








