Command Palette
Search for a command to run...
Shandong University Develops an Interpretable Deep Learning Algorithm RetroExplainer to Identify the Retrosynthetic Route of Organic Compounds in 4 Steps

Retrosynthesis aims to find a series of suitable reactants to efficiently synthesize the target product. This is an important method to solve the organic synthesis route and is also the simplest and most basic method for designing organic synthesis routes.
Early retrosynthesis research relied on programming, and this work was later taken over by AI. However, existing retrosynthesis methods focus on single-step retrosynthesis, have poor interpretability, and cannot take into account both short-range and long-range information of molecules, which limits their performance.
To this end, Wei Leyi from Shandong University and Zou Quan's research group from the University of Electronic Science and Technology of China jointly developed RetroExplainer. This explainable deep learning algorithm can identify the retrosynthetic route of organic compounds in 4 steps and give easily available reactants. RetroExplainer is expected to provide a powerful tool for retrosynthetic research in organic chemistry.
Author | Xuecai
Editor | Sanyang
Retrosynthesis in organic chemistry aims to find a series of suitable reactants to efficiently synthesize the target product.This process is an indispensable basic work in computer-aided synthesis.
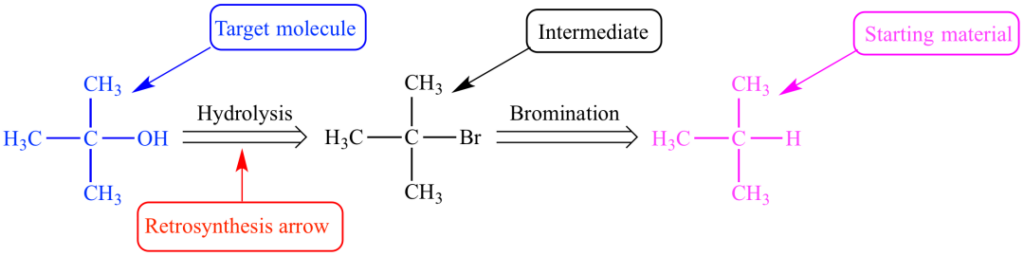
Figure 1: Retrosynthetic route of tert-butyl alcohol
In the 1960s,Corey et al. attempted to perform retrosynthetic analysis via programming, and developed the Organic Chemical Simulation Synthesis (OCSS) software. However,As the amount of data increases, this work is quickly taken over by AIAmong them, the Deep Learning model is highly anticipated and has produced considerable results.
In early AI retrosynthesis research, researchers often worked backward from products to reactants based on reaction templates, i.e., template-based retrosynthesis.Among them, molecular fingerprints based on multi-layer perceptrons are often used for product encoding and template selection.
Then,Researchers began to explore template-free and semi-template synthesis methods, mainly including:
1. Sequence-based retrosynthesis;
2. Diagram-based retrosynthesis.
The main difference between the two lies in the representation of molecules. The former uses linearized strings to represent molecules, such as the SMILES specification, while the latter uses molecular graph models to represent molecules, mainly including the prediction of reaction centers (RC) and the completion of synthons.
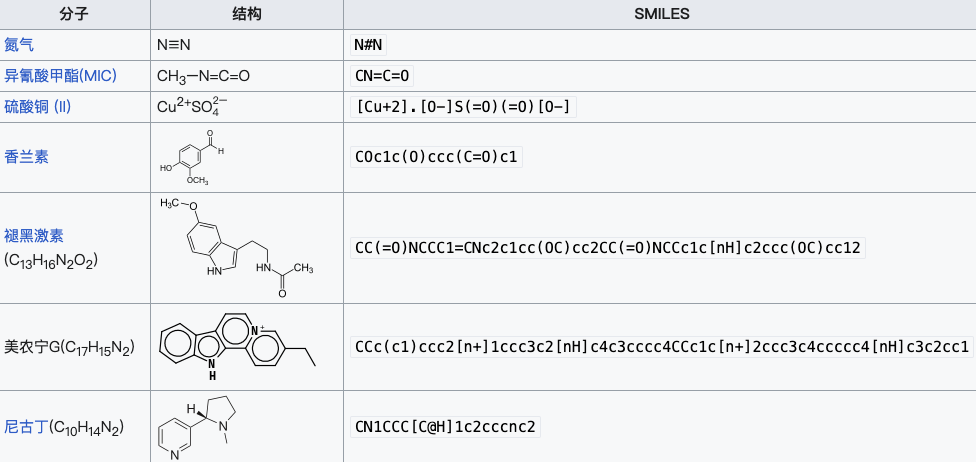
Figure 2: SMILES expressions of some substances
Although the existing retrosynthetic methods have made significant progress,However, there are still three endogenous problems:
1.Sequence-based retrosynthesis misses molecular information, while graph-based retrosynthesis ignores sequence information and long-range features of moleculesBoth methods are limited in feature learning and it is difficult to improve performance;
2.Deep learning-based retrosynthesis methods have poor interpretabilityAlthough template-based retrosynthesis can provide an easy-to-understand synthetic route, the decision-making mechanism of the algorithm is still vague, and the repeatability and feasibility of the model need to be considered;
3.Existing methods mostly focus on single-step retrosynthesisThis method seems to give reasonable reactants, but these reactants may be difficult to purchase or require complicated post-processing. Therefore, multi-step retrosynthesis may be more meaningful in actual chemical synthesis.
to this end,Wei Leyi from Shandong University and Zou Quan's research group from the University of Electronic Science and Technology of China jointly developed RetroExplainer . This algorithm can make retrosynthesis predictions based on deep learning, while taking into account the interpretability and feasibility of the algorithm. RetroExplainer outperformed other algorithms in almost 12 benchmark datasets, and 86.91% of the reactions in the proposed synthesis routes were verified by the literature. This result has been published in "Nature Communications".

This result has been published in Nature Communications.
Paper link:
https://www.nature.com/articles/s41467-023-41698-5
Follow the official account and reply "retrosynthesis" to get the full paper PDF
Experimental procedures
Algorithm construction:Module + Subgrid
The entire retrosynthetic analysis process includes four steps: molecular graphic encoding, multi-task learning, decision-making, and multi-step synthesis route prediction.
RetroExplainer mainly consists of four modules: Multi-sensory Multi-scale Graph Transformer (MSMS-GT), Dynamic Adaptive Multi-task Learning (DAMT), Explainable Decision Module and Route Prediction Module.
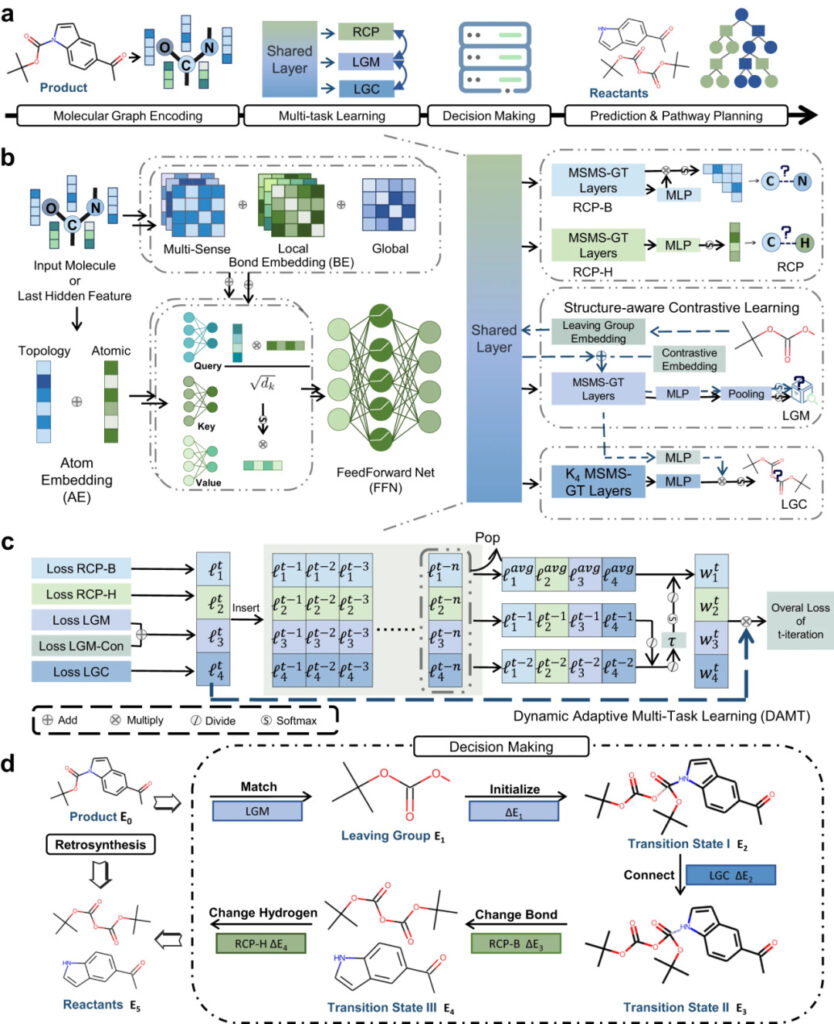
Figure 3: Schematic diagram of RetroExplainer and its modules
a: RetroExplainer process diagram;
b: MSMS-GT architecture;
c: Schematic diagram of DAMT algorithm;
d: Decision-making process similar to the reaction mechanism.
MSMS-GT captures important chemical information through chemical bond embedding and atomic topological embedding. The encoded information is fused into molecular vectors through a multi-head attention mechanism.
In the DAMT module, molecular information is simultaneously input into the Reaction Center Prediction (RCP), Leaving Group Match (LGM), and Leaving Group Connect (LGC) subgrids.
RCP identifies changes in chemical bonds and the number of hydrogen atoms adjacent to an atom, LGM matches the leaving group in the product to that in the database, and LGC links the leaving group to the product residue.
The decision module converts the product into the reactant based on the five retrosynthetic actions and the energy score (E) of the decision curve, and reversely simulates the molecular assembly process.
Finally, a heuristic tree search algorithm is used to find an efficient product synthesis route while ensuring the availability of reactants.
Performance comparison:USPTO Benchmark Dataset
To verify the performance of RetroExplainer, the researchers compared it with 21 other retrosynthesis algorithms based on chemical reactions included in the United States Patent and Trademark Office (USPTO), with the evaluation indicator being top-k accuracy.
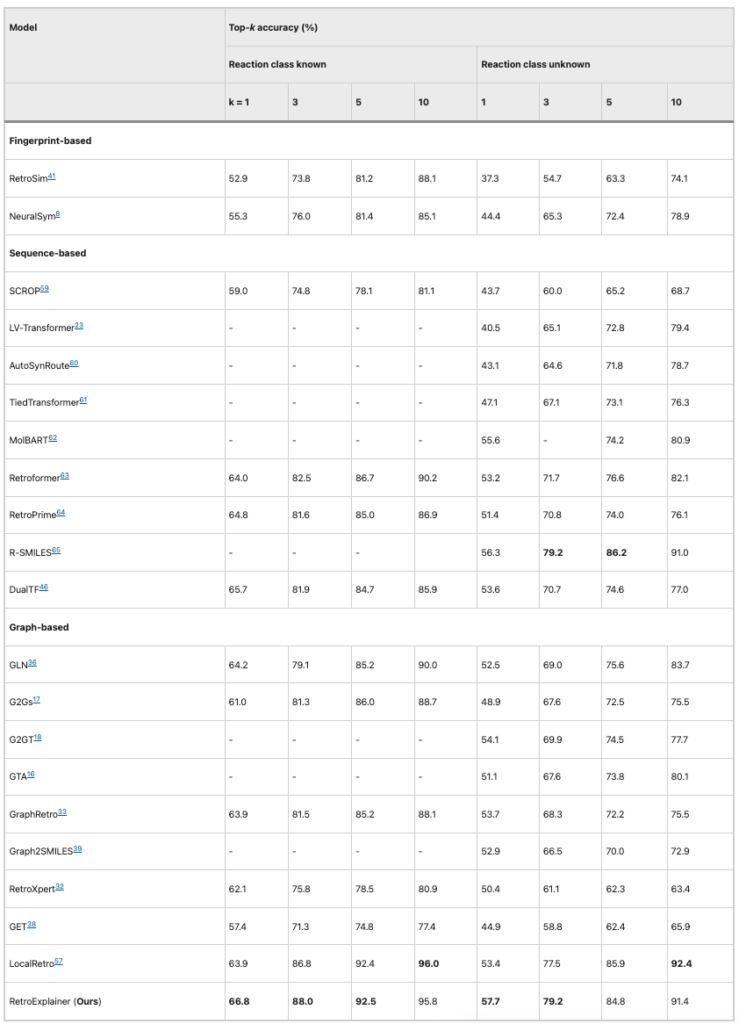
Table 1: Performance comparison of RetroExplainer and other algorithms (USPTO-50K)
It can be seen that based on the USPTO-50K dataset, among the 8 evaluation indicators,RetroExplainer outperforms other algorithms in 5 aspects and ranks first in average accuracyAlthough RetroExplainer is inferior to LocalRetro in top-10 accuracy, the gap between the two is only 1%.
To eliminate the impact of similar molecules, the researchers repartitioned the data using Tanimoto Similarity and compared it with the two most accurate algorithms, R-SMILE and LocalRetro.
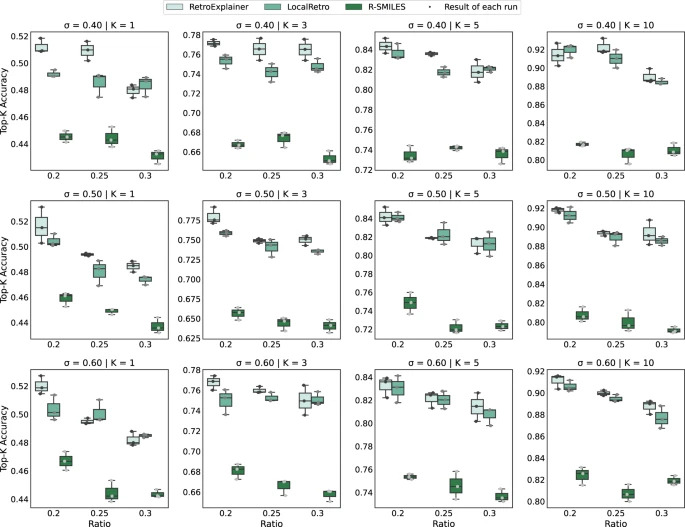
Figure 4: Performance comparison of RetroExplainer, R-SMILES and LocalRetro on different datasets
As can be seen from the results, RetroExplainer performs better in most datasets, reflecting its stability and adaptability.
Subsequently, the researchers compared the performance of the algorithms on the larger USPTO-MIT and USPTO-FULL datasets. RetroExplainer outperformed other algorithms in all indicators, and the gap between them and other algorithms was even greater.This shows that RetroExplainer has greater potential in large-scale data analysis.
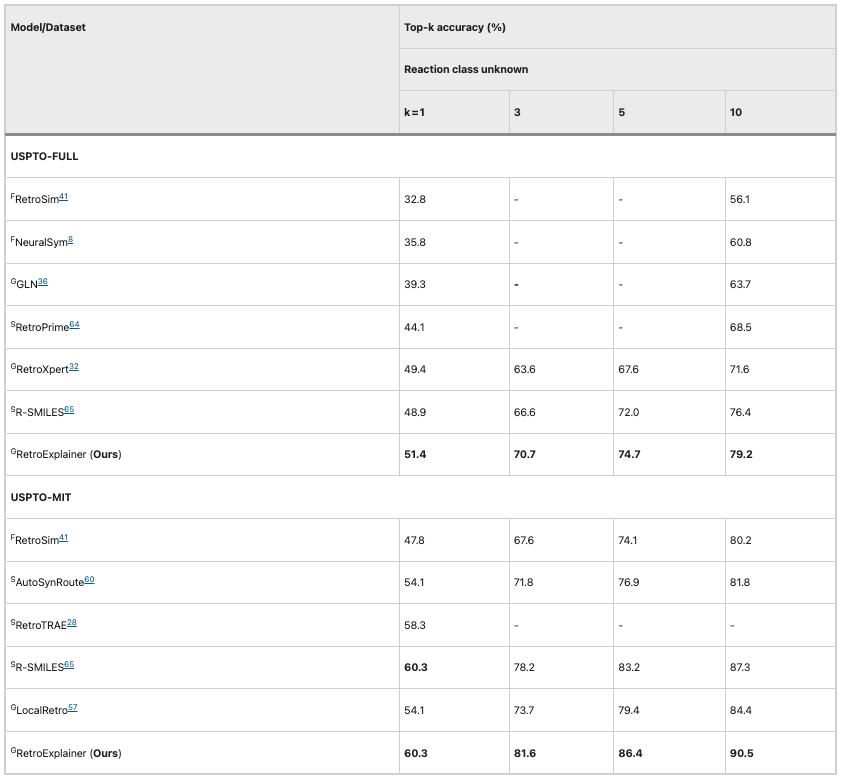
Table 2: Performance comparison of RetroExplainer and other algorithms (USPTO-MIT and USPTO-FULL)
Explainability:Decision Visualization
Inspired by the bimolecular nucleophilic substitution reaction (SN2), the researchers designed an interpretable retrosynthetic prediction process based on deep learning-guided molecular assembly. The decision-making process includes 6 stages: original product (P), leaving group matching (S-LGM), initialization (IT), leaving group connection (S-LGC), reaction center chemical bond change (S-RCP), and hydrogen atom number change (HC).
Based on the contribution of each stage to the final decision, the DAMT subgrid generates an energy score (E) for each stage.
The specific process is as follows:
1. In the P stage, E of each stage is initialized to 0;
2. In the S-LGM stage, the leaving group is selected based on the predicted probability of the LGM module;
3. Add the E of the leaving group selected in the S-LGM stage to the response event probability predicted by the RCP and LGM modules to obtain the energy of the IT stage;
4. In the S-LGC and S-RCP phases, based on the dynamic programming algorithm, all possible nodes in the search tree are expanded. Events with probabilities greater than a preset threshold are selected, and E is fixed;
5. Adjust the number of hydrogen atoms and formal charge of each atom to ensure that the resulting molecular diagram conforms to the valence rules and calculate the final E.
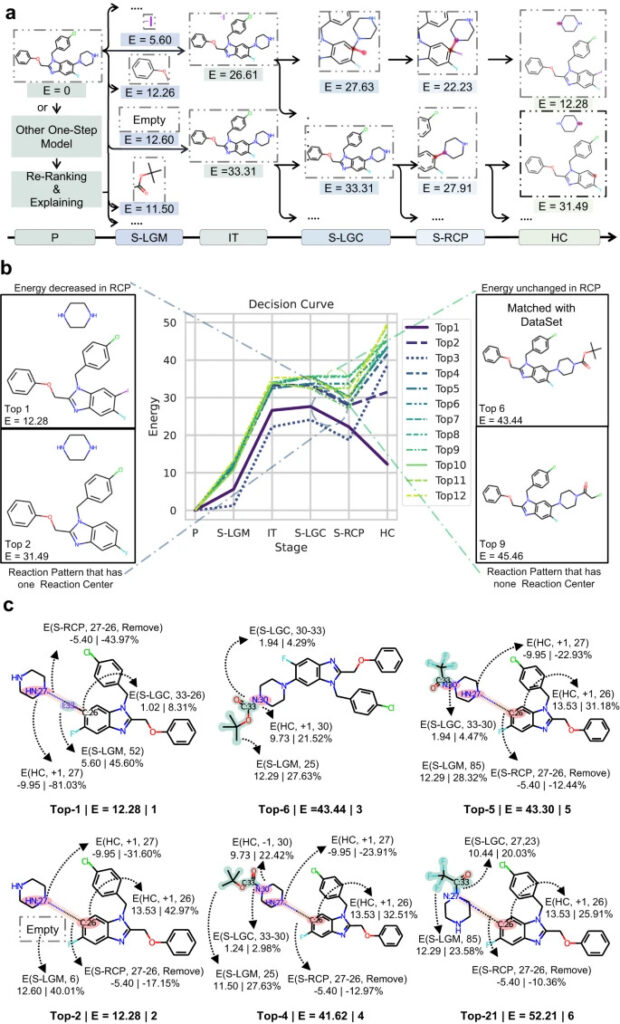
Figure 5: RetroExplainer’s decision-making process
a: RetroExplainer’s search path for two prediction results;
b: Decision curves of the top 12 predicted routes;
c: 6 structural change processes representing the synthetic routes.
By drawing a decision curve based on the change of E, we can analyze the decision process of RetroExplainer and find out the prediction errors of RetroExplainer.
As shown in the figure, the correct synthetic route for the product should be the deprotection reaction of the amine, but RetroExplainer ranked it 6th, and the 1st was the CN coupling reaction. Analysis found that RetroExplainer tended to increase the number of hydrogen atoms of the amine in the HC stage, which led to this difference.This indicates that RetroExplainer may make the same misjudgment of molecules with similar structures at the HC stage.
By comparing the reactions of RetroExplainer ranked 1st and 2nd,Researchers found that E may be related to the difficulty of the responseAlthough the connection between I:33 and C:26 in reaction 1 is not conducive to energy reduction, connecting a hydrogen atom at C:26 requires 13 times the energy of the previous reaction. At the same time, the introduction of I:33 weakens the selectivity problem faced by the CN coupling reaction.
at the same time,Steric hindrance can also affect the prediction results of RetroExplainerComparing the reactions ranked 4th and 21st, their molecular structures are the same, but the leaving groups are attached to the symmetrical N, resulting in differences in E.
Path Planning:Multi-step predicted synthetic routes
To improve the practicality of RetroExplainer's predictions, the researchers combined it with the Retro algorithm, replacing the latter's single-step predictions with multi-step predictions.
Taking the bronchodilator Protokylol as an example, RetroExplainer designed a four-step synthesis route for this product. Subsequently, the researchers conducted a literature survey on the four-step reaction to explore its feasibility.
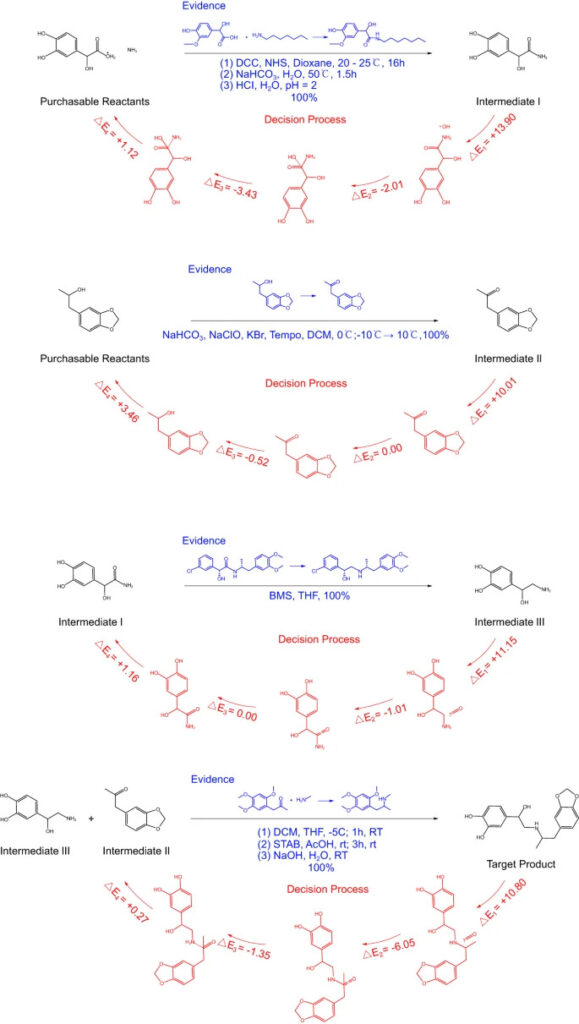
Figure 6: RetroExplainer's 4-step synthesis of protosan
The blue text in the figure is a similar response recorded in the reference, and the red part is the decision-making process of RetroExplainer.
Although many reactions did not find exactly the same reference, they found similar high-yield reactions.RetroExplainer designed 176 experiments for 101 cases, of which 153 could find similar reactions in SciFinder.
The above results show that RetroExplainer is better than other existing algorithms in predicting retrosynthesis. At the same time, RetroExplainer's decision-making is transparent and explainable, and it can plan reactions in multiple steps, making it more feasible. RetroExplainer is expected to provide a powerful tool for retrosynthesis research in organic chemistry.
Performance vs. explainability, the contradiction of AI
Explainability is a key factor in applying AI in various scenarios. With the continuous development of AI in industries such as driverless driving, medical diagnosis, and financial insurance, the decision-making process of AI has become increasingly important and is facing more and more practical, social, and even legal issues.
At the same time, explainability can help users understand, maintain and use AI, and discover and understand new concepts in AI application areas. Explainability also reflects the feasibility of the results and tells users that the benefits of this decision are the greatest.
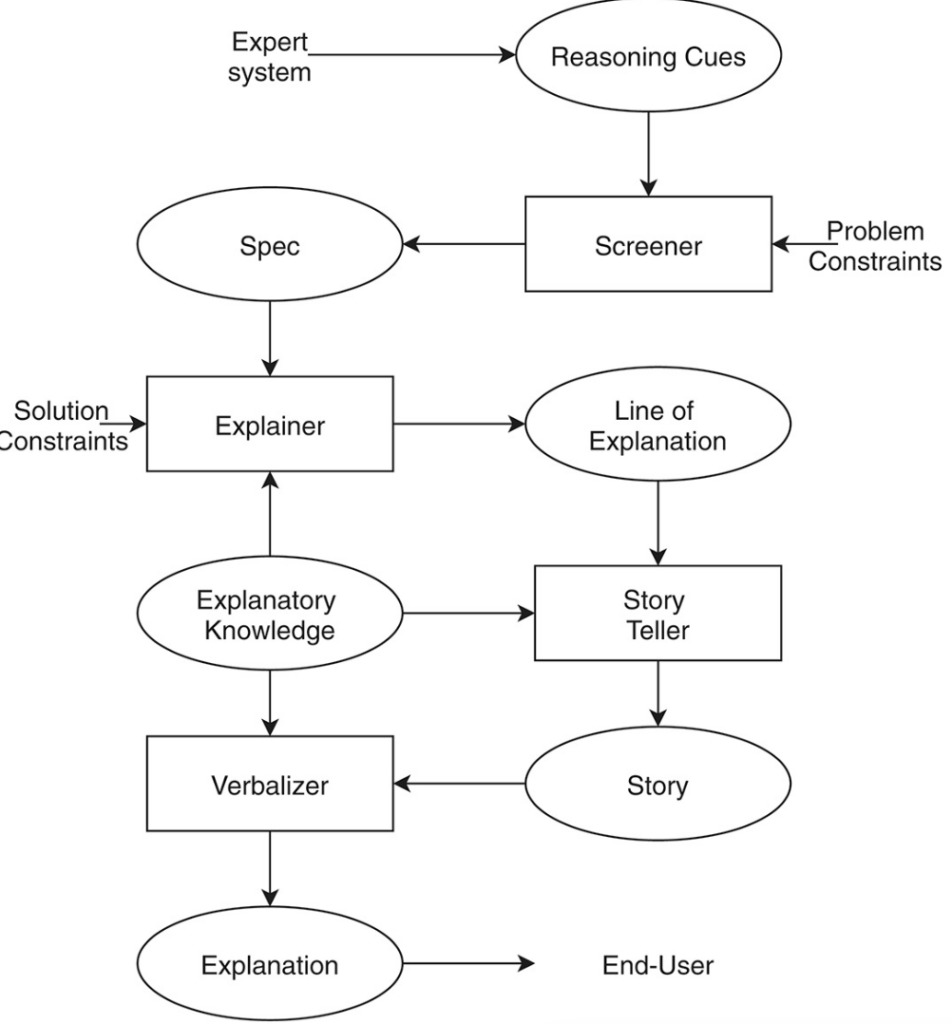
Figure 7: Explanation phase of the problem-solving process
However,Model performance and model interpretability are a big problem for ScienceAIIf the model has good performance and good cross-test set robustness, then high-dimensional deep features may work better, but they do not have any physical meaning, which is what we often call "The interpretability of scientific research is generally poor".
On the contrary, if well-explained features are used, although it is physically very interpretable, the actual model performance will have a large dependence on the data, and the model performance will decline if the data set is changed.
There is still no good way to unify the contradiction between the two, but in this study, the researchers visualized the decision-making process of AI step by step., allowing users to clearly understand the score changes of various prediction results at each stage, understand the decision-making process of AI, and also facilitate developers to optimize the model.
With the continuous development of explainable AI, people's understanding of AI will become deeper and the decision-making process of AI will become easier to understand.In the future, the interaction between humans and machines will continue to increase, the threshold for interaction will be further lowered, and AI will be used in more scenarios, making life more convenient and intelligent.
Reference Links:
[1]http://www.chem.ucla.edu/~harding/IGOC/R/retrosynthesis.html
[2] https://zh.wikipedia.org/zh-cn/Simplified molecular linear input specification
[3]https://wires.onlinelibrary.wiley.com/doi/10.1002/widm.1391








