Command Palette
Search for a command to run...
Tutorial Zur Einzelzell-Transkriptomsequenzierung Einzelner Proben: Qualitätskontrolle, Clustering, (differenzielle) Gendarstellung
Datum
Größe
2.42 GB
Paper-URL
Tutorial zur Einzelzell-Transkriptomsequenzierungsanalyse
Die in dieser Studie verwendeten Beispieldaten stammen aus einem Artikel, der im Juli 2024 in Nature Medicine veröffentlicht wurde. Personalisierter Neoantigen-Impfstoff und Pembrolizumab bei fortgeschrittenem hepatozellulärem Karzinom: eine Phase-1/2-Studie Einzelzellsequenzierungsdaten von mononukleären Zellen des peripheren Blutes von Patient 6 (Yarchoan M et al. Nat Med. 2024;30:1044–1053.).
Einführung in die Einzelzellsequenzierung
Die Einzelzellsequenzierung ist eine hochentwickelte Biotechnologie, die es Wissenschaftlern ermöglicht, die Genexpression auf der Ebene einzelner Zellen eingehend zu analysieren. Der Prozess beginnt typischerweise mit einer Gewebeprobe, die zunächst in einzelne Zellen zerlegt wird, um eine Einzelzellsuspension zu bilden. Anschließend wurde jede Zelle mithilfe der Wasser-in-Öl-Technik einzeln eingekapselt, um sicherzustellen, dass sie während der nachfolgenden Analyse unabhängig blieb.
Bei der Einzelzellsequenzierung wird das mRNA-Molekül jeder Zelle mit einer einzigartigen molekularen Kennung markiert, ein Prozess, der als UMI-Markierung (Unique Molecular Identifier) bezeichnet wird. Gleichzeitig wird jeder Zelle ein gleicher Barcode-Tag zugewiesen, der während der Datenanalysephase zur Rückverfolgung der Quelle jeder Zelle verwendet wird. Auf diese Weise können Forscher die mRNA-Informationen in jeder Zelle präzise messen und so den Genexpressionstatus jeder Zelle verstehen.
1. Barcode-Funktion: Zellinformationen aufzeichnen
2. Anwendung von UMI in der Einzelzellsequenzierung: Durch die Bereitstellung einer eindeutigen Kennung für jedes mRNA-Molekül wird nicht nur die Genauigkeit der Genexpressionmessung verbessert, sondern auch die Fehler reduziert, die während der PCR-Amplifikation auftreten können, wodurch die Zuverlässigkeit und Effektivität des gesamten Sequenzierungsprozesses verbessert wird.
Grundlegender Prozess
Der Prozess der Einzelzell-RNA-Sequenzierung (scRNA-seq) umfasst hauptsächlich die Isolierung und Erfassung einzelner Zellen, die Zelllyse, die reverse Transkription (Umwandlung von RNA in cDNA), die cDNA-Amplifikation und die Bibliotheksvorbereitung
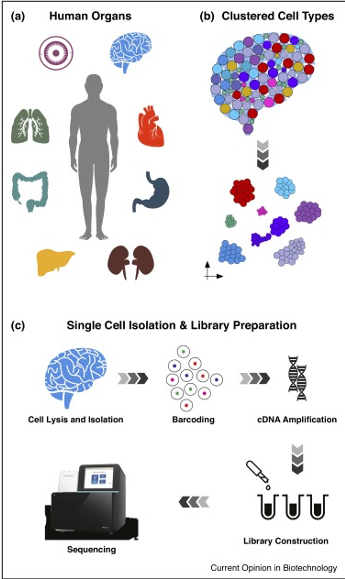
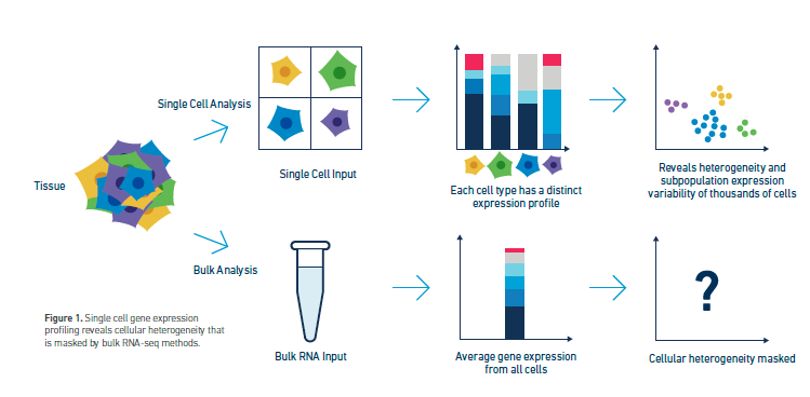
Bildquelle: Kulkarni, A., et al. (2019). „Mehr als Masse: Ein Überblick über Methoden und Anwendungen der Einzelzell-Transkriptomik„ Curr Opin Biotechnol 58: 129-136
Schritte ausführen
Nun beginnen wir mit dem praktischen Teil des Tutorials.
Starten Sie den Container und laden Sie Daten hoch
1. Starten Sie den Container
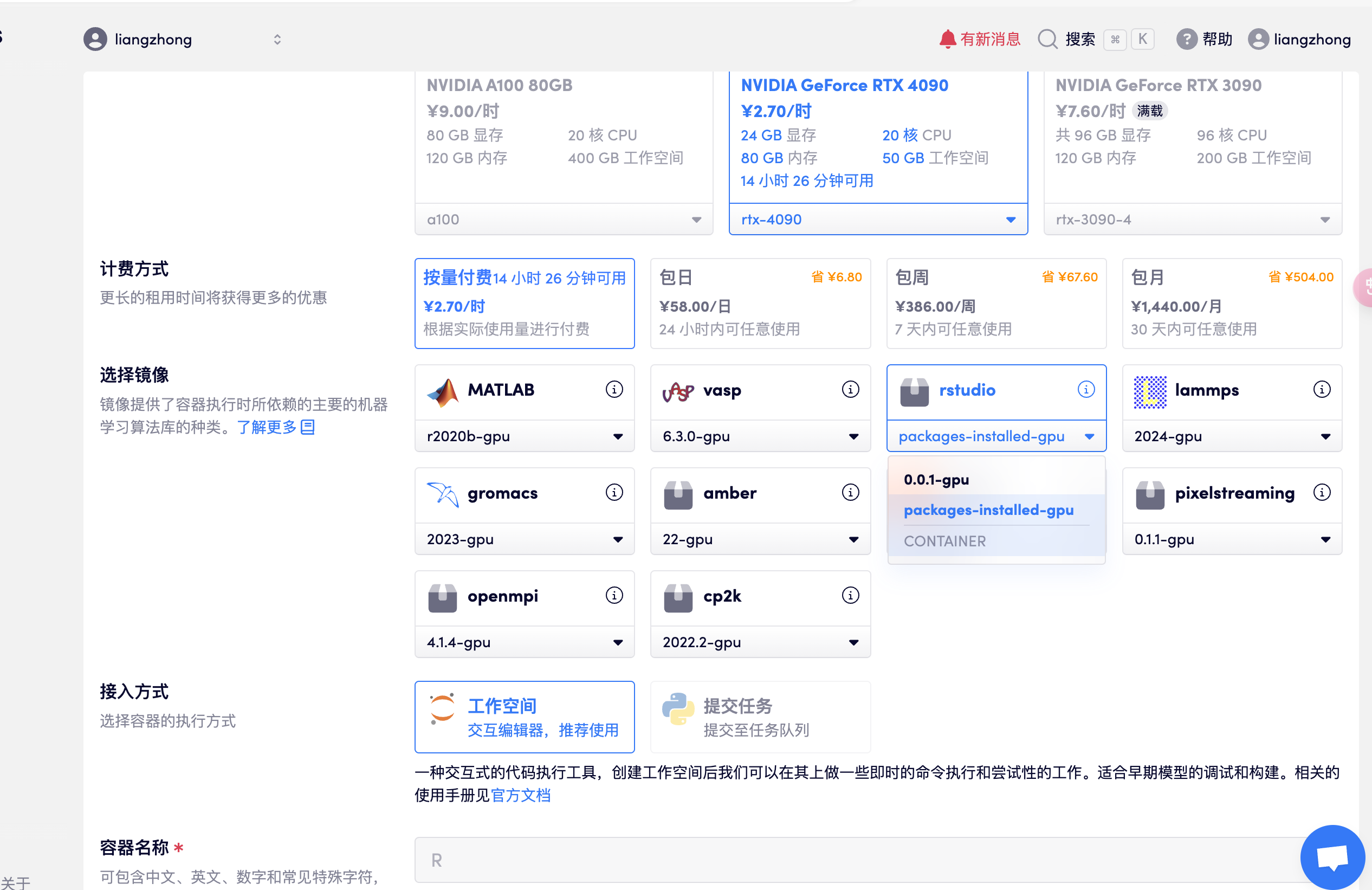
Erstellen Sie einen Container, öffnen Sie RStudio, wählen Sie die im Paket installierte GPU-Version aus und öffnen Sie nach der Ressourcenzuweisung direkt die API-Adresse, um RStudio aufzurufen (für diesen Schritt ist eine Echtnamenauthentifizierung erforderlich), wie in der folgenden Abbildung dargestellt:
- Hinweis: Nach dem Aufrufen der Software lauten Konto und Passwort: rstudio
2. Daten hochladen
Laden Sie die vorbereiteten Daten vorab hoch (die Daten können im Datenordner des Arbeitsverzeichnisses heruntergeladen und verwendet werden):
(1) Rohdaten einzelner Zellen, sequenziert mit Illumina NextSeq 500: barcodes.tsv.gz, features.tsv.gz, matrix.mtx.gz
(2) Referenzdaten für die Zellannotation: pbmc_multimodal.h5seurat, ref_Human_all.RData
#存放的目录如下
#此处可新建自定义文件夹,指定输入输出文件目录,本教程以 output 为例
rstudio@****(用户名)-bs5z6fbf81vrmain:~/home/output/data$ ls
barcodes.tsv.gz features.tsv.gz matrix.mtx.gz
rstudio@****(用户名)-bs5z6fbf81vr-main:~/home/output/reference_annotation$ ls
pbmc_multimodal.h5seurat ref_Human_all.RDataGeben Sie anschließend den Code gemäß den Schritten des Tutorials Schritt für Schritt in R Studio ein.
#报错改为英文
Sys.setenv(LANGUAGE = "en")
options(stringsAsFactors = FALSE)
#清空所有数据
rm(list=ls())
set.seed(100)#!!!设置随机数,保证后续实验的可重复性1. Richten Sie das Seurat-Objekt ein
# I. Setup -----------------------------------------------------------------------
# ## a. Load libraries ----------------------------------------------------------------------
```{r libraries}
library(Seurat)
library(SeuratObject)
library(sctransform)
library(patchwork)
library(Seurat)
library(SeuratData)
library(patchwork)
library(SeuratData)
library(ggplot2)
library(tidyverse)
library(glmGamPoi)
library(tools)
library(dplyr)
library(patchwork)2. Daten laden
# II. Load data-----------------------------------------------------------------------
### a. Load data-----------------------------------------------------------------
getwd()#查看当前工作目录
setwd("~/home/output/results")
#修改工作目录,此处可根据自定义文件夹,指定输入输出文件目录,本教程以 output 为例
pbmc.data <- Read10X(data.dir ="~/home/output/data")#设置 10x 原始数据的工作目录,读取
seurat <- CreateSeuratObject(counts = pbmc.data, project = "pbmc", min.cells = 3, min.features = 200)#创建 seurat 对象
seurat
An object of class Seurat
16356 features across 10066 samples within 1 assay
Active assay: RNA (16356 features, 0 variable features)
1 layer present: counts
#该样本有 10066 个细胞 ,10066 个单细胞的 RNA 表达数据,测序了 16356 个基因
#counts: 通常表示原始的基因表达计数数据
colnames([email protected])
[1] "orig.ident" "nCount_RNA" "nFeature_RNA" "percent.mt" "RNA_snn_res.0.1" "seurat_clusters"
[7] "celltype"
Die pbmc.data-Daten werden in der folgenden Abbildung mit Lesevorgängen angezeigt, Zeilennamen sind Gene und Spaltennamen sind Zell-Barcodes.
pbmc.data[c("CD3D", "CD8A", "PTPRC"), 1:30] 3 x 30 spärliche Matrix der Klasse "dgCMatrix" [[Unterdrückung der 30 Spaltennamen 'AAACCTGAGCGAAGGG-1', 'AAACCTGAGGCCCTTG-1', 'AAACCTGAGGGTTTCT-1' … ]]
CD3D 2 . 3 6 4 1 1 3 . 6 3 4 2 1 2 1 . 2 2 5 2 3 . 6 . 4 3 CD8A . 6 . . . 6 PTPRC 1 2 2 2 3 5 3 3 3 1 3 . 2 4 1 . 1 . 2 9 . 3 4 . 1 2 . 2 1 4
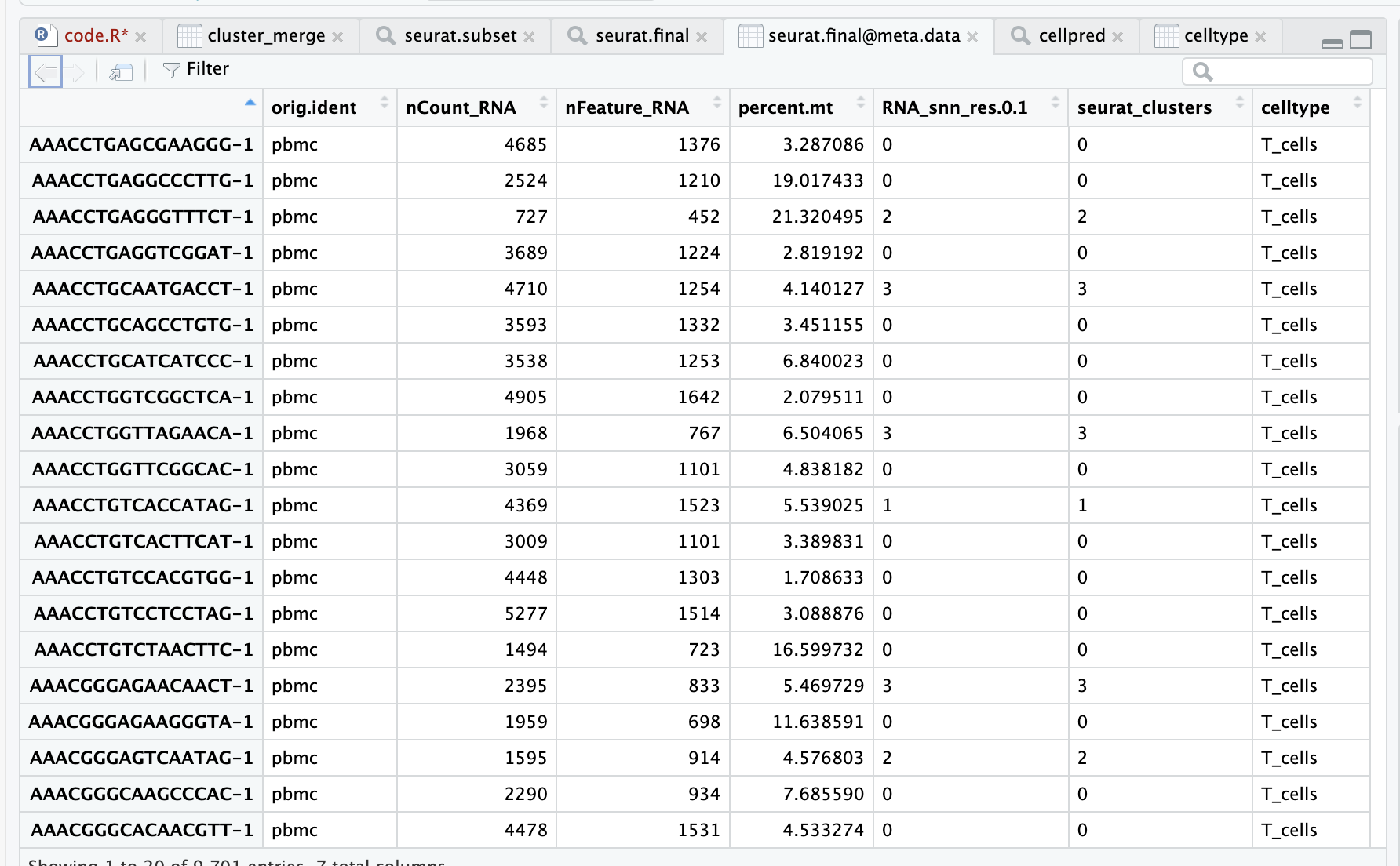
- Beschreibung der Tabelleninformationen:
- Der Zeilenname lautet Barcodes
- orig.ident ist die Probeninformation,
- nCount_RNA ist die gesamte Genexpression
- nFeature_RNA ist die Anzahl der Gene
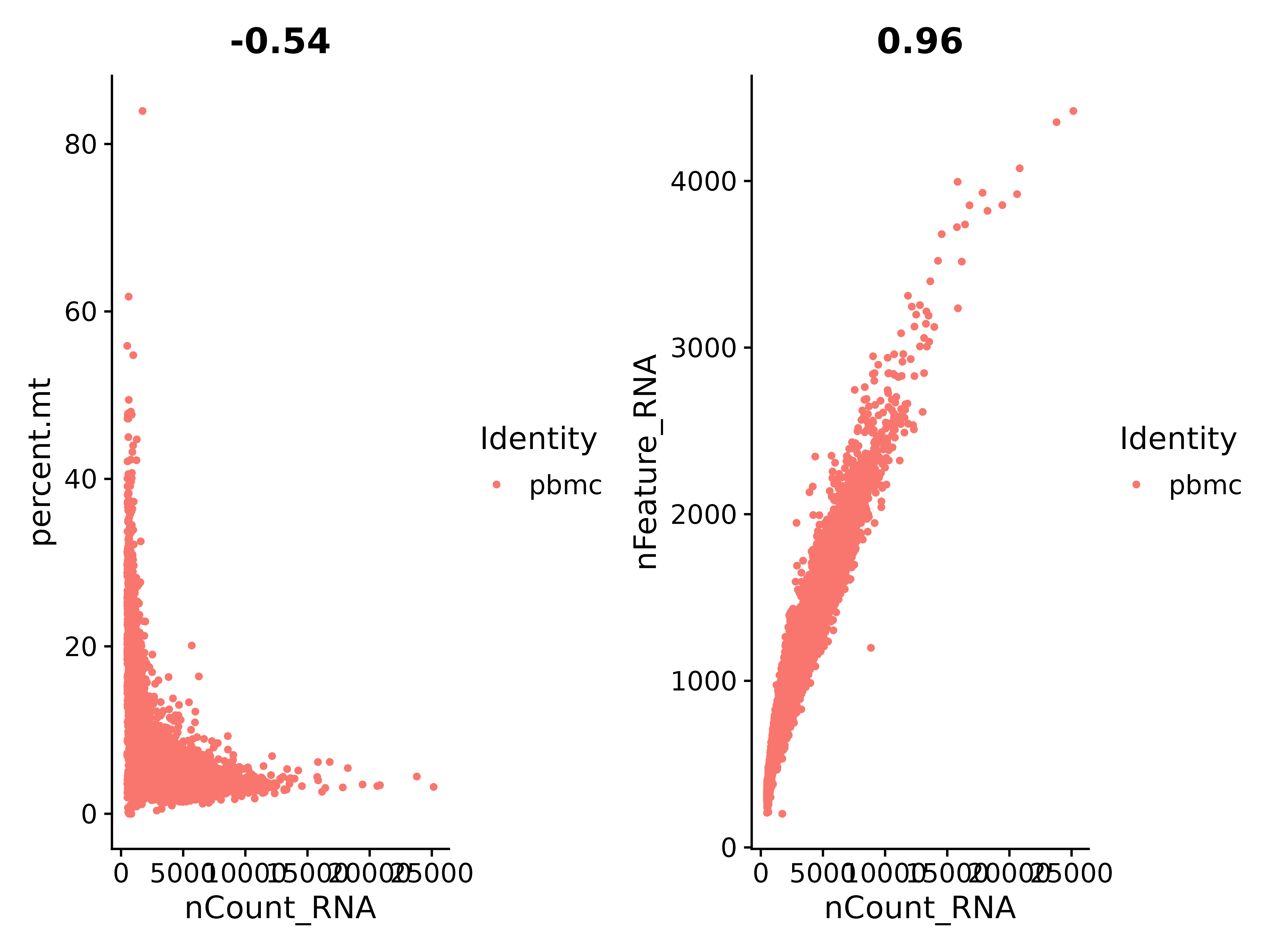
Überprüfen wir anhand der obigen Abbildung das Verhältnis zwischen nCount_RNA und percent.mt (mitochondriales Gen):
Es ist ersichtlich, dass nCount_RNA und nFeature_RNA in einem positiven Verhältnis stehen, mit einem Koeffizienten von 0,96, und die Korrelation sehr gut ist.
Tipps: Alle Gene, die mit MT- beginnen, sind mitochondriale Gene.
3. Standard-Vorbehandlung
Die folgenden Schritte decken den Standard-Vorverarbeitungs-Workflow für scRNA-seq-Daten in Seurat ab. Zu diesen Prozessen gehören das Auswählen und Filtern von Zellen auf der Grundlage von QC-Metriken, die Normalisierung und Skalierung von Daten sowie das Erkennen stark variabler Merkmale.
Seurat kann QC-Metriken anzeigen und Zellen basierend auf benutzerdefinierten Kriterien filtern.
# III. Pre-processing----------------------------------------------------------------
### a. Plots for percent mito, nFeature---------------------------------------------
```{r preprocessing}
# The [[ operator can add columns to object metadata. This is a great place to stash QC stats
seurat <-PercentageFeatureSet(seurat, pattern = "^MT-", col.name = "percent.mt")
# Use FeatureScatter is typically used to visualize feature-feature relationships, but can be used
# for anything calculated by the object, i.e. columns in object metadata, PC scores etc.
plot1 <- FeatureScatter(seurat, feature1 = "nCount_RNA", feature2 = "percent.mt",)
plot2 <- FeatureScatter(seurat, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
plot1 + plot2
ggsave("质控_1.png", plot1 + plot2, width = 8, height = 6, dpi=600)
#plot3 <- FeatureScatter(seurat, feature1 = "nCount_RNA", feature2 = "percent.ribo")
#plot3
Als Nächstes visualisieren wir die QC-Metriken und verwenden sie zum Filtern von Zellen.
Betrachten Sie den Anteil mitochondrialer Gene und verwenden Sie Violindiagramme zur Visualisierung von QC-Indikatoren
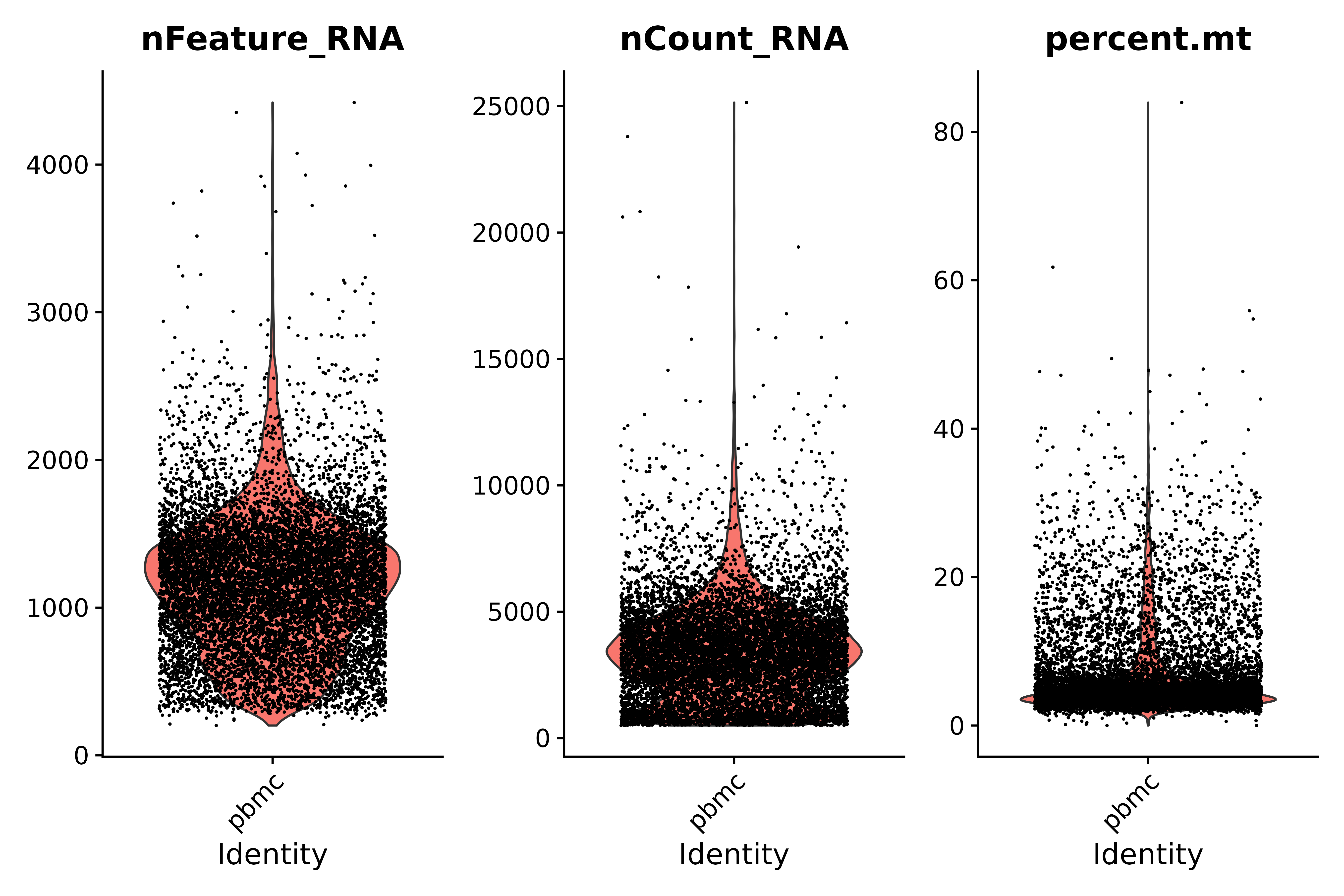
- Anweisungen zur Qualitätskontrolle:
- Minderwertige Zellen oder leere Tröpfchen weisen typischerweise sehr wenige Gene auf, wobei nFeature_RNA > 200 und nCount_RNA > 500 gilt.
- Diploide oder polyploide Zellen können ungewöhnlich hohe Genanzahlen aufweisen; laut unserer Grafik gilt: nFeature_RNA < 2500 & nCount_RNA > 500
- Minderwertige/sterbende Zellen weisen in der Regel eine ausgedehnte mitochondriale Hyperplasie auf, percent.mt < 25
# Visualize QC metrics as a violin plot
vln1 <- VlnPlot(seurat, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"#,"percent.ribo"
),ncol=3)
vln1
ggsave("质控_2.png", vln1, width = 9, height = 6, dpi=600)seurat.subset <- subset(seurat, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & nCount_RNA > 500 & nCount_RNA < 10000 &percent.mt < 25)
vln3 <- VlnPlot(seurat.subset, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
vln3
ggsave("质控_3.png", vln3, width = 9, height = 6, dpi=600)
pdf("qc_plots.pdf");plot1 + plot2;vln1;vln3;dev.off()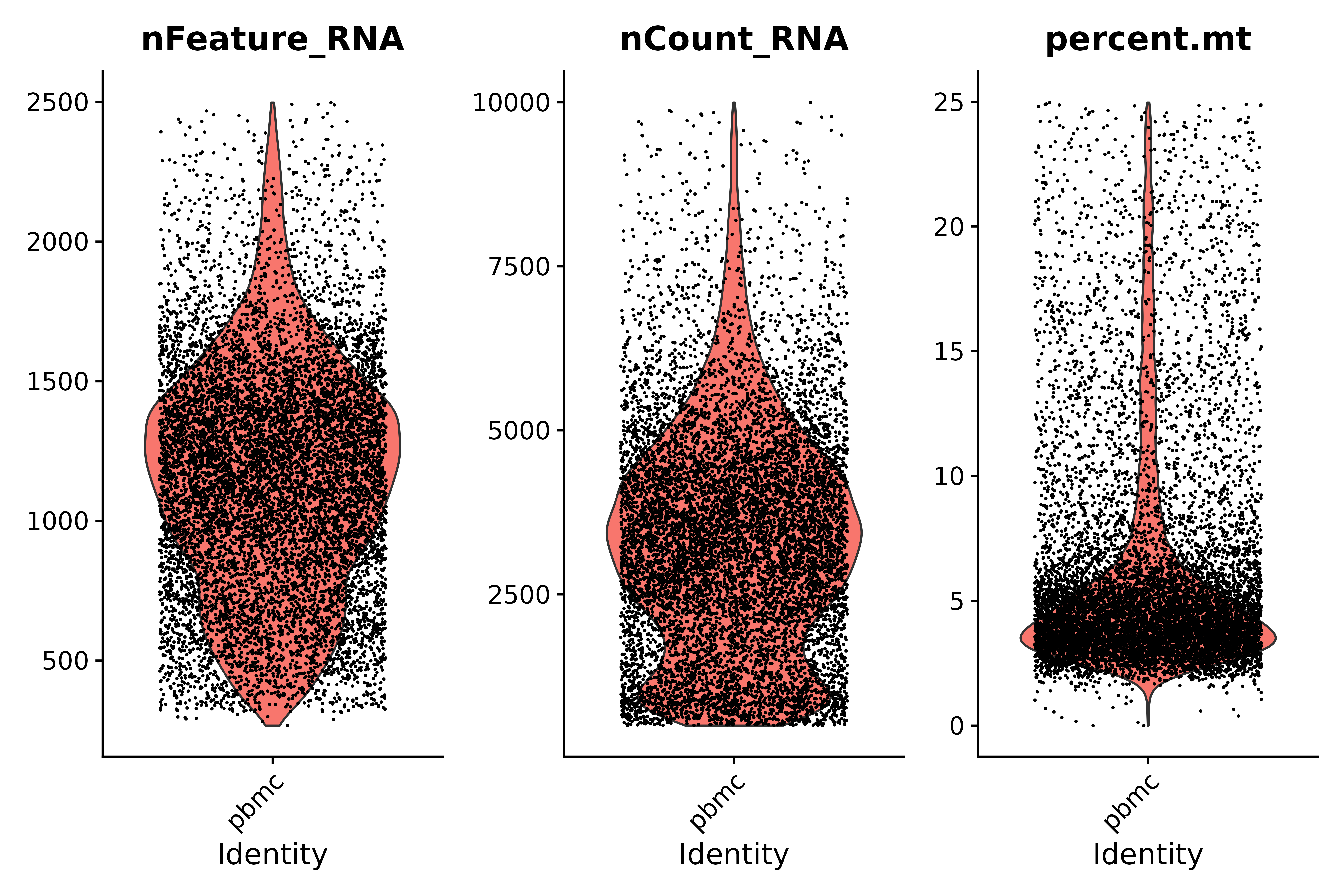
4. Normalisieren Sie die Daten und identifizieren Sie stark variable Merkmale (Merkmalsauswahl).
Bei der Analyse von Einzelzell-Transkriptomdaten führen wir zunächst eine Datennormalisierung durch, um sicherzustellen, dass die Genexpressionsniveaus zwischen verschiedenen Zellen effektiv verglichen werden können.
1. Datennormalisierung
- Datennormalisierung: Wir teilen zunächst das Expressionsniveau (die Anzahl) eines bestimmten Gens in jeder Zelle durch das Gesamtexpressionsniveau (die Gesamtanzahl) der Zelle. Dieser Schritt dient dazu, die Unterschiede in der Sequenzierungstiefe zwischen Zellen zu beseitigen, sodass die Genexpressionsniveaus verschiedener Zellen auf derselben Basis verglichen werden können.
- Skalierungsanpassung: Der normalisierte Expressionswert wird dann mit einem Skalierungsfaktor, hier 10000, multipliziert, um den Genexpressionswert für die nachfolgende Analyse auf eine geeignetere Größenordnung anzupassen.
- Logarithmische Transformation: Abschließend führten wir eine logarithmische Transformation der angepassten Expressionswerte durch. Dies trug dazu bei, die Varianz weiter zu stabilisieren und die Unterschiede zwischen hoch- und niedrigexprimierten Genen deutlicher zu machen. Dies legte den Grundstein für nachfolgende Analyseschritte.
- Normalisierung: Durch die oben genannten Schritte haben wir die Genexpressionswerte auf das Niveau von jeweils 10.000 UMIs normalisiert, was dazu beiträgt, die Auswirkungen von Unterschieden in der Sequenzierungstiefe zwischen Zellen zu eliminieren.
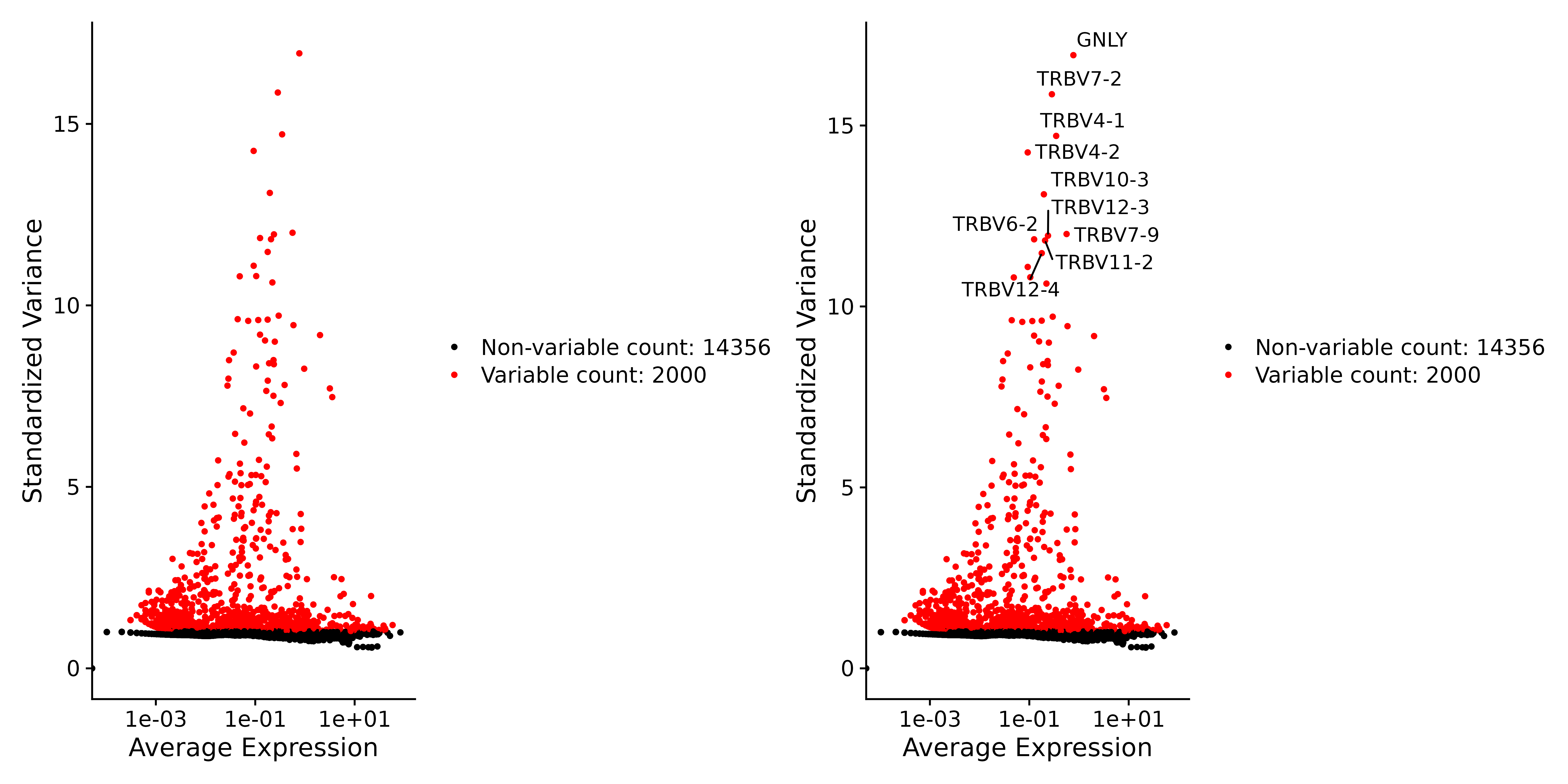
2. Als nächstes suchen wir nach Mutationsmerkmalen:
Nach der Normalisierung und Standardisierung identifizierten wir hypervariable Gene durch Analyse der Variabilität der Genexpression. Diese Gene weisen in unterschiedlichen Zellen deutlich unterschiedliche Expressionsniveaus auf und können in einigen Zellen stark exprimiert sein, in anderen jedoch auf niedrigerem Niveau.
# IV. Normalize Data----------------------------------------------------------------
seurat.subset <- NormalizeData(seurat.subset, normalization.method = "LogNormalize", scale.factor = 10000)
seurat.subset <- FindVariableFeatures(seurat.subset, selection.method = "vst", nfeatures = 2000)
# Identify the 10 most highly variable genes
top10 <- head(VariableFeatures(seurat.subset), 10)
# plot variable features with and without labels
plot3 <- VariableFeaturePlot(seurat.subset)
plot4 <- LabelPoints(plot = plot3, points = top10, repel = TRUE)
ggsave("VariableFeaturePlot_4.png", plot3 + plot4, width = 12, height = 6, dpi=600)
5. Daten skalieren und PCA ausführen
Bei der Analyse von Einzelzelltranskriptomdaten können Datenskalierung und PCA die Dimension der Daten effektiv reduzieren, die Hauptquellen der Variation zwischen Zellen extrahieren und wichtige Informationen zum Verständnis der Zellheterogenität und -funktion liefern.
1. Daten skalieren: Die Datenskalierung ist eine lineare Transformation, deren Ziel darin besteht, Genexpressionsdaten in eine Standardnormalverteilung umzuwandeln, d. h. der Mittelwert der Daten beträgt 0 und die Standardabweichung 1. Diese Transformation trägt dazu bei, die Unterschiede zwischen den Expressionsniveaus verschiedener Gene zu verringern und die Daten stärker zu normalisieren. Durch die Skalierung kann der Einfluss von Extremwerten oder Ausreißern in den Daten reduziert werden, wodurch die Datenverteilung gleichmäßiger wird und ein stabilerer Input für nachfolgende Analyseschritte bereitgestellt wird. Dieser Schritt dient auch der Vorbereitung für die Hauptkomponentenanalyse (PCA), da die PCA bei standardisierten Daten bessere Ergebnisse liefert.
2. Führen Sie PCA aus:
- PCA ist eine statistische Methode, die Daten in ein neues Koordinatensystem überträgt, indem die orthogonalen Achsen so transformiert werden, dass die erste größte Varianz einer beliebigen Projektion der Daten auf der ersten Koordinate liegt (die sogenannte erste Hauptkomponente), die zweitgrößte Varianz auf der zweiten Koordinate und so weiter.
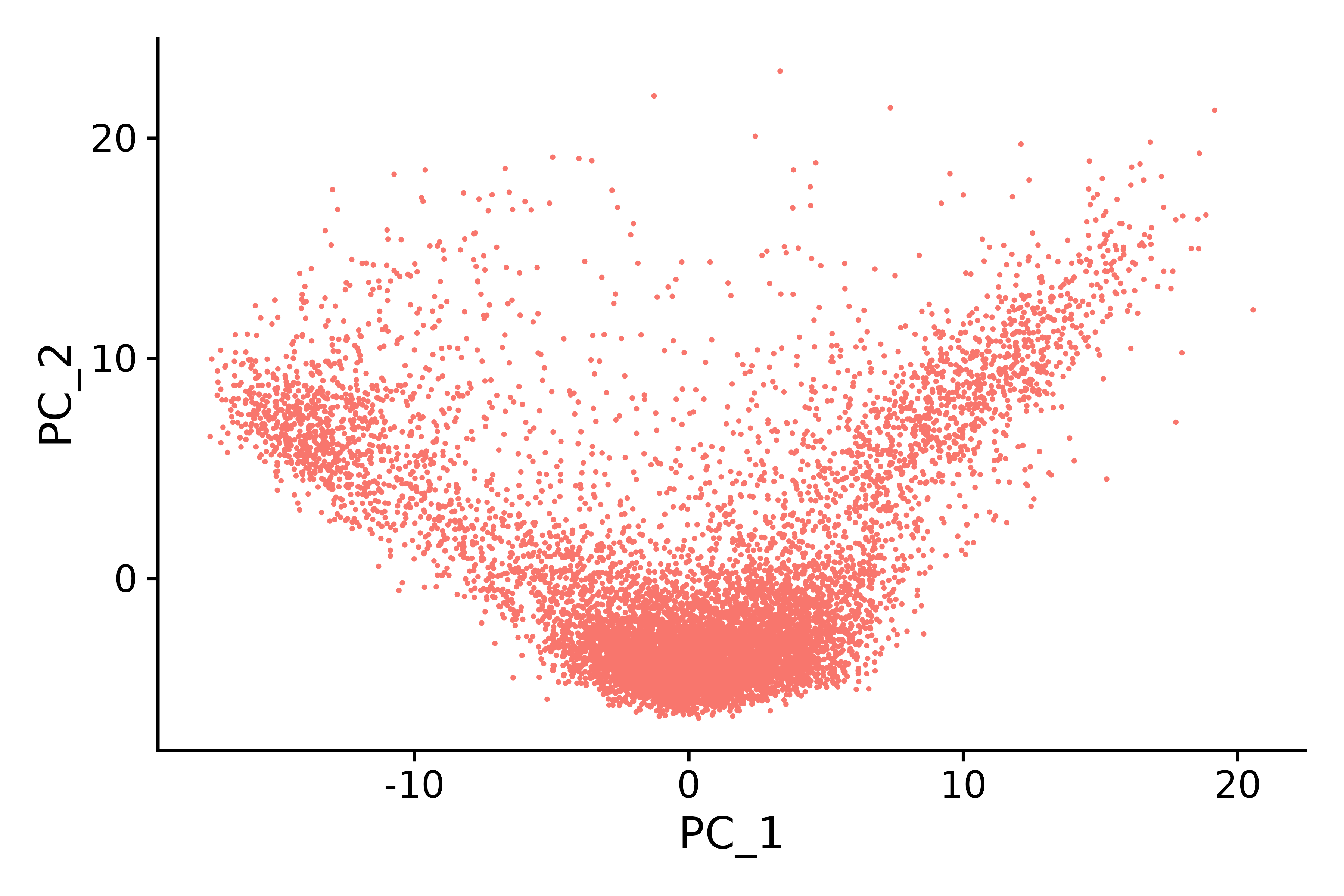
- Bei der Einzelzell-Transkriptomanalyse wird PCA verwendet, um einzelne Zellen auf der Grundlage von Unterschieden in der Genexpression zu trennen. Jede Zelle kann als Punkt in einem hochdimensionalen Raum betrachtet werden, und PCA stellt diese Punkte in einem niedrigdimensionalen Raum neu dar, wobei möglichst viele Variationsinformationen erhalten bleiben.
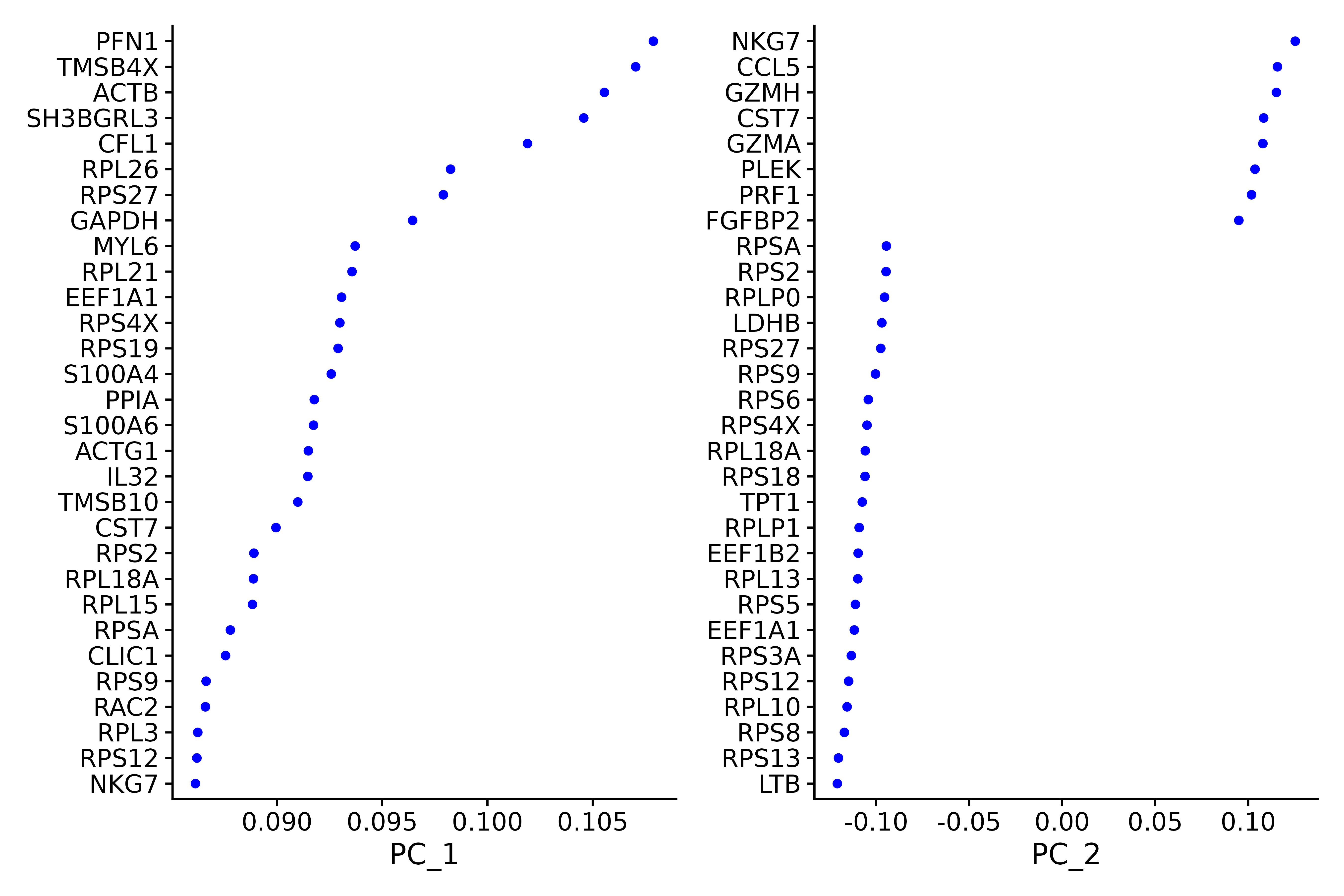
- Jede Hauptkomponente (PCA) stellt im Wesentlichen ein „Metamerkmal“ der Zelle dar, d. h. eine Hauptvariationsrichtung der Zelle. In den PCA-Ergebnissen gilt: Je weiter vorne die Hauptkomponente liegt, desto größer ist ihr Gewicht in der Datenvariation, was bedeutet, dass sie mehr Informationen enthält.
- Durch PCA können die hochdimensionalen Expressionsinformationen von Zellen in mehrere wichtige Hauptkomponenten komprimiert werden, die für nachfolgende Clusteranalysen, Visualisierungen oder andere nachgelagerte Analysen verwendet werden können.
all.genes <- rownames(seurat.subset)
seurat.subset <- ScaleData(seurat.subset, features = all.genes)
seurat.subset <- RunPCA(seurat.subset, features = VariableFeatures(object = seurat))##### Examine and visualize PCA results a few different ways-------------
print(seurat.subset[["pca"]], dims = 1:5, nfeatures = 5)
p1 <- VizDimLoadings(seurat.subset, dims = 1:2, reduction = "pca")
p1
ggsave("VizDimLoadings_5.png", p1, width = 9, height = 6, dpi=600)
p2 <- DimPlot(seurat.subset, reduction = "pca") + NoLegend()
p2
ggsave("pca_6.png", p2, width = 6, height = 4, dpi=600)
p3 <- DimHeatmap(seurat.subset, dims = 1, cells = 500, balanced = TRUE)
p3
ggsave("pca_1dims.png", p3, width = 6, height = 4, dpi=600)
p4 <- DimHeatmap(seurat.subset, dims = 1:15, cells = 500, balanced = TRUE)
p4
ggsave("pca_15dims.png", p4, width = 10, height = 10, dpi=600)
p5 <- ElbowPlot(seurat.subset)
p5
ggsave("ElbowPlot.png", p5, width = 10, height = 10, dpi=600)
DimHeatmap() ist ein leistungsstarkes Datenanalysetool, mit dem Forscher wichtige Heterogenitätsquellen in einem Datensatz einfach identifizieren und untersuchen können. Nach der Durchführung einer Hauptkomponentenanalyse (PCA) ist DimHeatmap() besonders hilfreich bei der Bestimmung, welche Hauptkomponenten (PCs) für die nachfolgende eingehende Analyse einbezogen werden sollten. Bei diesem Prozess werden Zellen und Merkmale anhand von PCA-Werten eingestuft, um die Genauigkeit und Relevanz der Analyse sicherzustellen.
Darüber hinaus ermöglicht DimHeatmap() dem Benutzer, eine bestimmte Anzahl von Zellen festzulegen, um gezielt „extreme“ Zellen darzustellen, die an beiden Enden des Datenspektrums extreme Eigenschaften aufweisen. Dieser Ansatz verbessert die Plot-Effizienz bei der Arbeit mit großen Datensätzen erheblich, da er sich auf die wichtigsten Teile der Daten konzentriert und so den gesamten Visualisierungsprozess beschleunigt.
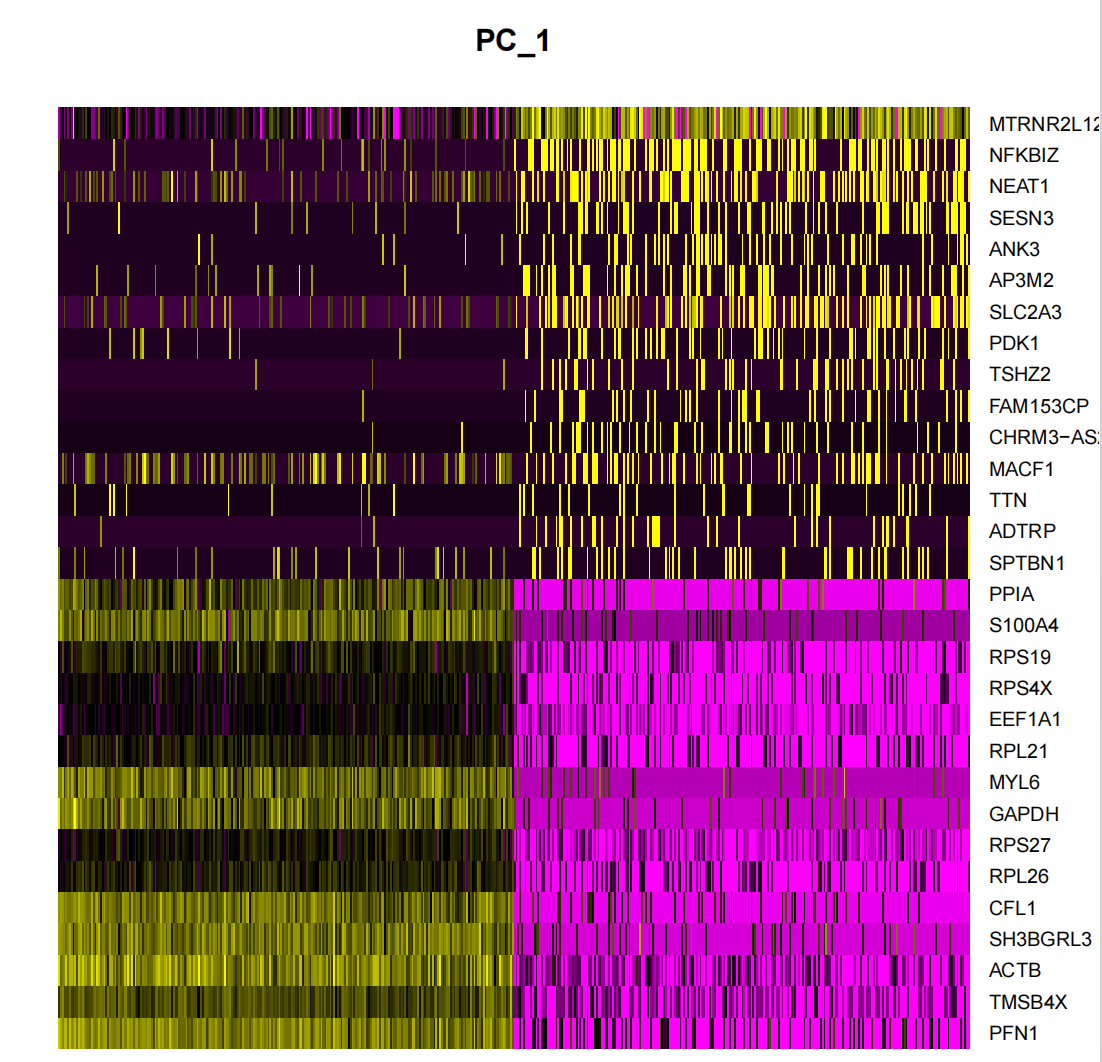

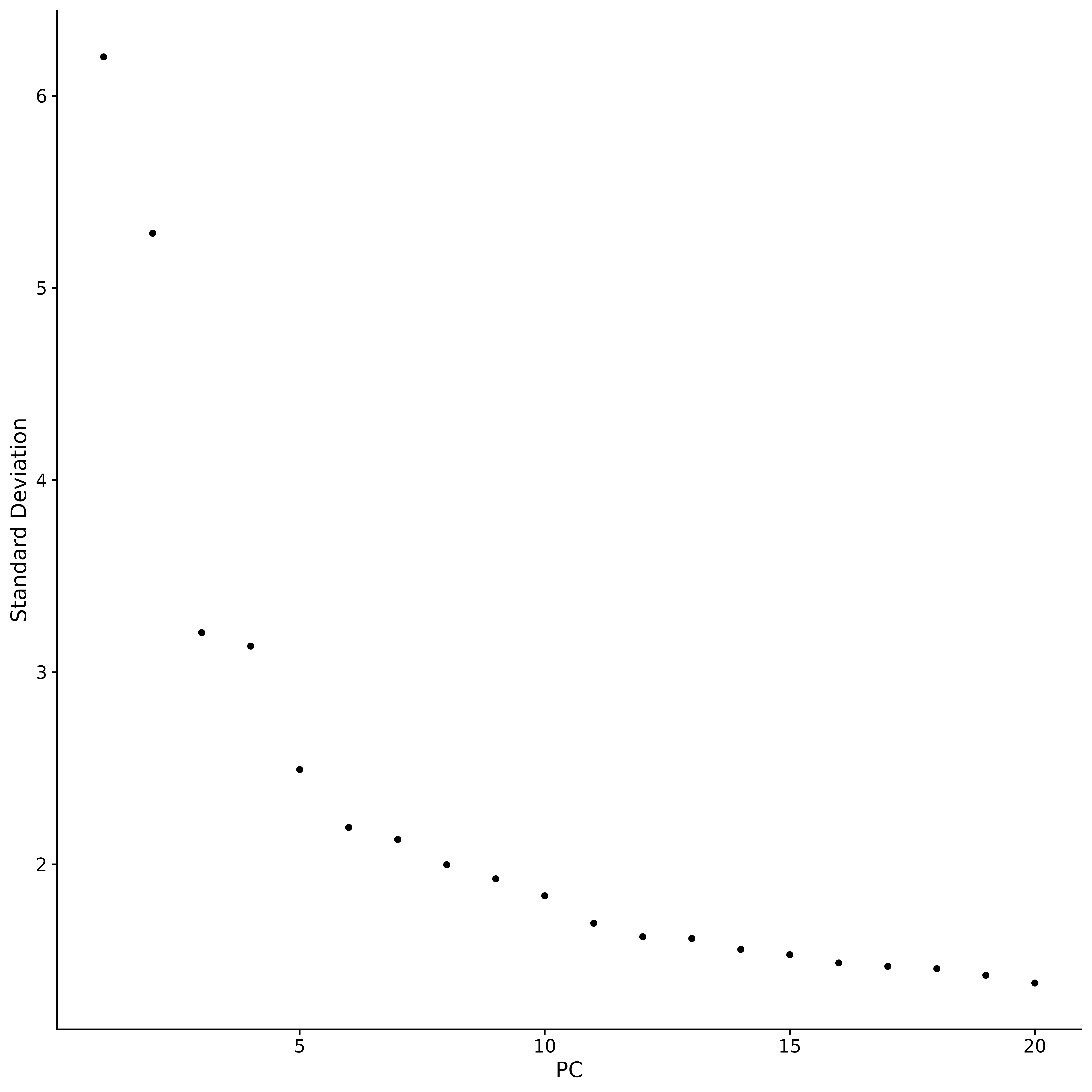
Bei der Hauptkomponentenanalyse (PCA) ist es üblich, mithilfe des „Elbow Plot“ zu bestimmen, wie viele Hauptkomponenten für die nachfolgende Analyse beibehalten werden sollen. Die Darstellung zeigt, dass der Großteil des wahren Signals in den ersten 17 Hauptkomponenten erfasst wird. Im Elbow-Plot suchen wir nach einem Wendepunkt, dem „Elbow“. Vor diesem Punkt verbessert jede zusätzliche Hauptkomponente die Aussagekraft der Daten erheblich, nach diesem Punkt beginnt jedoch der Nutzen durch das Hinzufügen von Hauptkomponenten abzunehmen.Dies zeigt an, dass ein Gleichgewichtszustand erreicht wurde.. Basierend auf den Analyseergebnissen des Ellenbogendiagramms haben wir die ersten 17 Hauptkomponenten für die anschließende Analyse ausgewählt. Denn das Hinzufügen weiterer Hauptkomponenten verbessert die Unterscheidung der Daten nicht wesentlich, führt aber zu einer unnötigen Erhöhung des Rechenaufwands. Wenn wir die Anzahl der Hauptkomponenten weiter erhöhen, kann die Unterscheidung der Daten möglicherweise leicht verbessert werden, diese Verbesserung ist jedoch nicht signifikant und führt zu einem erheblichen Anstieg der Rechenkosten. Daher ist die Wahl von 17 Hauptkomponenten eine vernünftige Wahl, die ein Gleichgewicht zwischen Unterscheidungsvermögen und Rechenleistung herstellt.
pdf("dimreduction_plots.pdf")#保存为 pdf 文件
VizDimLoadings(seurat.subset, dims = 1:2, reduction = "pca")
DimPlot(seurat.subset, reduction = "pca")
DimHeatmap(seurat.subset, dims = 1, cells = 500, balanced = TRUE)
DimHeatmap(seurat.subset, dims = 1:15, cells = 500, balanced = TRUE)
ElbowPlot(seurat.subset)
dev.off()seurat.subset.dimReduction <- seurat.subset
seurat.subset.dimReduction <- FindNeighbors(seurat.subset.dimReduction, reduction="pca", dims = 1:17)
saveRDS(seurat.subset.dimReduction, file = "~/home/output/data/seurat_pre_FindClusters.rds")
# seurat.subset.dimReduction <- readRDS("~/home/output/data/seurat_pre_FindClusters.rds")6. Zellen clustern
### 3. Find clusters,聚类
```{r find_clusters}
seurat.final <- FindClusters(object = seurat.subset.dimReduction, resolution = 0.1)
head(Idents(seurat.final), 5)
seurat.final <- RunUMAP(seurat.final, reduction="pca", dims=1:17)
#Visualize with UMAP
p1<- DimPlot(seurat.final, reduction = "umap")
p1
ggsave("find_clusters.png", p1, width = 6, height =5 , dpi=600)
saveRDS(seurat.final, file = "~/home/output/data/seurat_final.rds")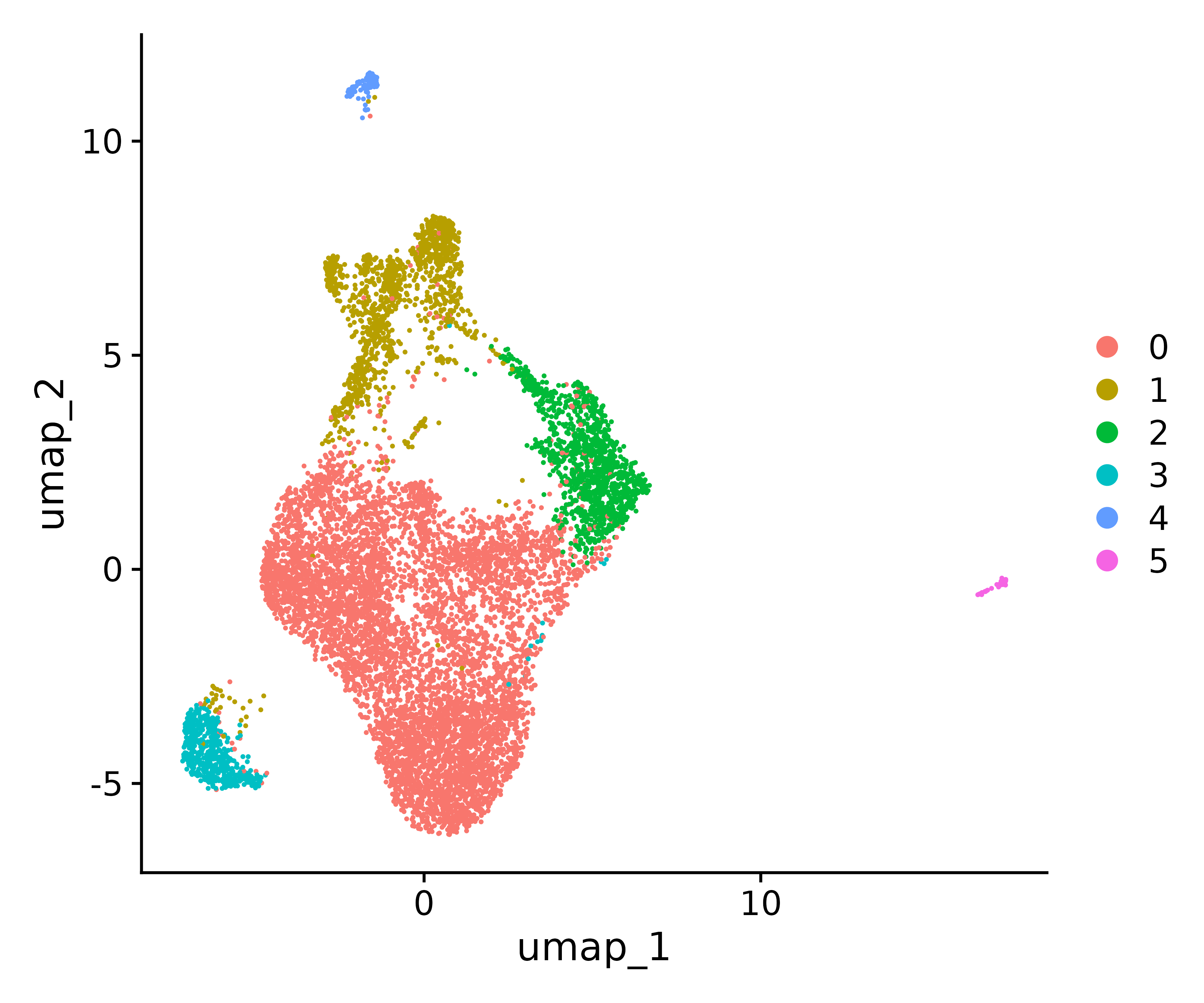
Visualisierung
Es gibt drei gängige Diagramme zur Anzeige der Genexpression in verschiedenen Clustern:
1. Heatmap 2. Blasenkarte 3. Featureplot
table(Idents(seurat.final))
# find markers between cluster1 and cluster 2
##用来找 cluster 之间的寻找差异表达特征
cluster2.markers <- FindMarkers(seurat.final, ident.1 = 1,ident.2 = 2)
head(cluster2.markers, n = 5)
# find markers for every cluster compared to all remaining cells, report only the positive ones
#找每个 cluster 的 marker 基因
pbmc.markers <- FindAllMarkers(seurat.final, only.pos = TRUE)
ClusterMarker_noRibo <- pbmc.markers[!grepl("^RP[SL]", pbmc.markers$gene, ignore.case = F),]
ClusterMarker_noRibo_noMito <- ClusterMarker_noRibo[!grepl("^MT-", ClusterMarker_noRibo$gene, ignore.case = F),]
###去除核糖体基因和线粒体基因的影响
ClusterMarker_noRibo_noMito %>%
group_by(cluster) %>%
dplyr::filter(avg_log2FC > 1) %>%
slice_head(n = 5) %>%
ungroup() -> top5
###展示每个 cluster 的 top5 的基因
heatmap <- DoHeatmap(seurat.final, features = top5$gene) + NoLegend()
ggsave("heatmap.png", heatmap, width = 10, height =5 , dpi=600) Diese Abbildung ist eine Heatmap, die Markergene zeigt
Diese Abbildung ist eine Heatmap, die Markergene zeigt
g =unique(top5$gene)
##定义 top5 的 gene 列,作为基因集 g,也可以自定义
p <- DotPlot(seurat.final,features=g)+
coord_flip() + # 翻转坐标轴
theme(
panel.grid = element_blank(), # 移除网格线
axis.text.x = element_text(angle = 0, hjust = 0.5, vjust = 0.5) # 旋转 x 轴标签
)
ggsave("allmarker_dotpot.png", p, width = 8, height =7 , dpi=600)
#气泡图展示 marker 基因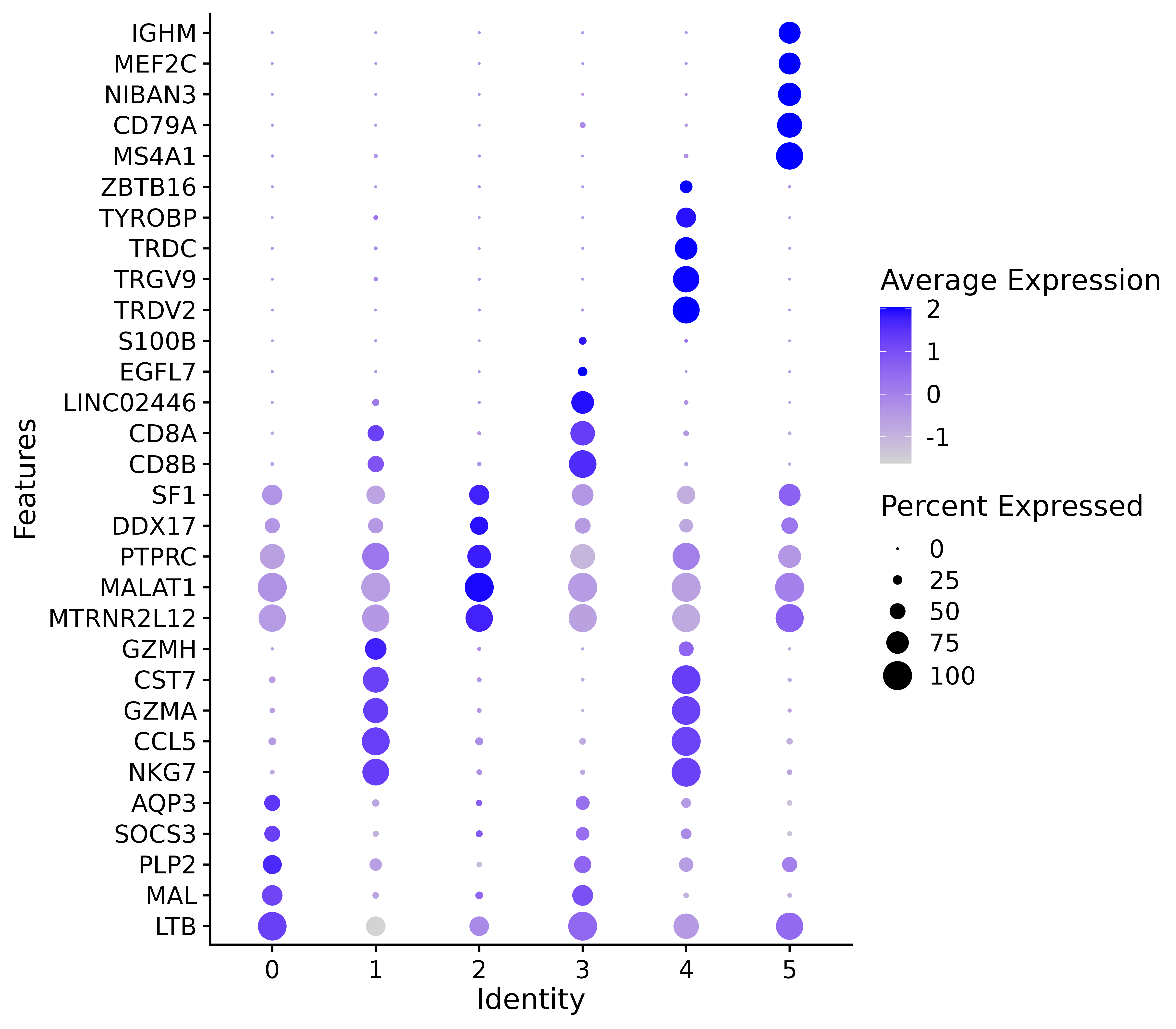
Diese Abbildung ist ein Blasendiagramm, das Markergene zeigt
# 加载 dplyr 包
library(dplyr)
# 提取 cluster 和 gene 列
cluster_gene_mapping <- top5 %>%
select(cluster, gene)
# 查看结果
head(cluster_gene_mapping,30)
write.csv(cluster_gene_mapping,"cluster_allmarkers_TOP5.csv", row.names = p <- FeaturePlot(seurat.final, features = g)
ggsave("allmarker_featurepot.png", p, width = 15, height =15 , dpi=600)
###查看自定义感兴趣的基因
p2 <- FeaturePlot(seurat.final, features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A", "FCGR3A", "LYZ", "PPBP",
"CD8A"))
ggsave("allmarker_featurepot2.png", p2, width = 15, height =15 , dpi=600)


7. Datenanalyse
In diesem Schritt wird beschrieben, wie Sie mithilfe spezifischer Markergene Zelltypen in der Transkriptomdatenanalyse einzelner Zellen bestimmen und definieren.
Vorausgesetzt, Sie haben den Zelltyp anhand von Top-Markergenen usw. bestimmt, können Sie den Identitätstyp definieren
0 1 2 3 4 5 „CD4 T“, „NK“, „Treg“, „CD8 T“, „γδ T-Zelle“, „B-Zelle“
(Die folgenden Kommentare dienen nur als Referenz und sind keine tatsächlichen Kommentare.)
new.cluster.ids <- c("CD4 T", "NK", "Treg", "CD8 T", "γδ T cell",
"B cell")
names(new.cluster.ids) <- levels(seurat.final)
seurat.final <- RenameIdents(seurat.final, new.cluster.ids)
p <- DimPlot(seurat.final, reduction = "umap", label = TRUE, pt.size = 0.5) + NoLegend()
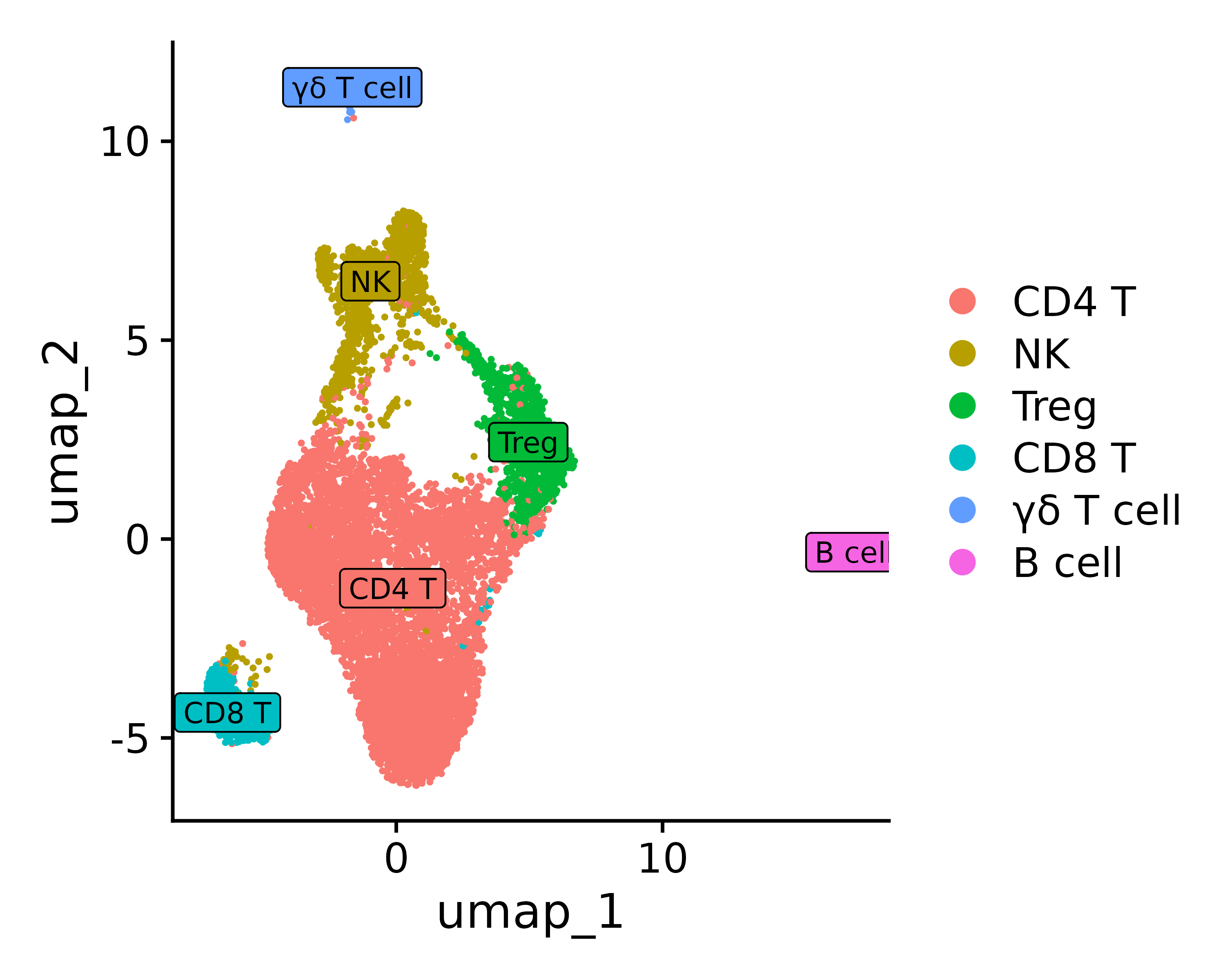
p3 = DimPlot(seurat.final, reduction = "umap", label = TRUE, label.size = 3, pt.size = .3, label.box = T)
ggsave("annotation_umap.png", p, width = 5, height =4 , dpi=600)
ggsave("annotation_umap2.png", p3, width = 5, height =4 , dpi=600)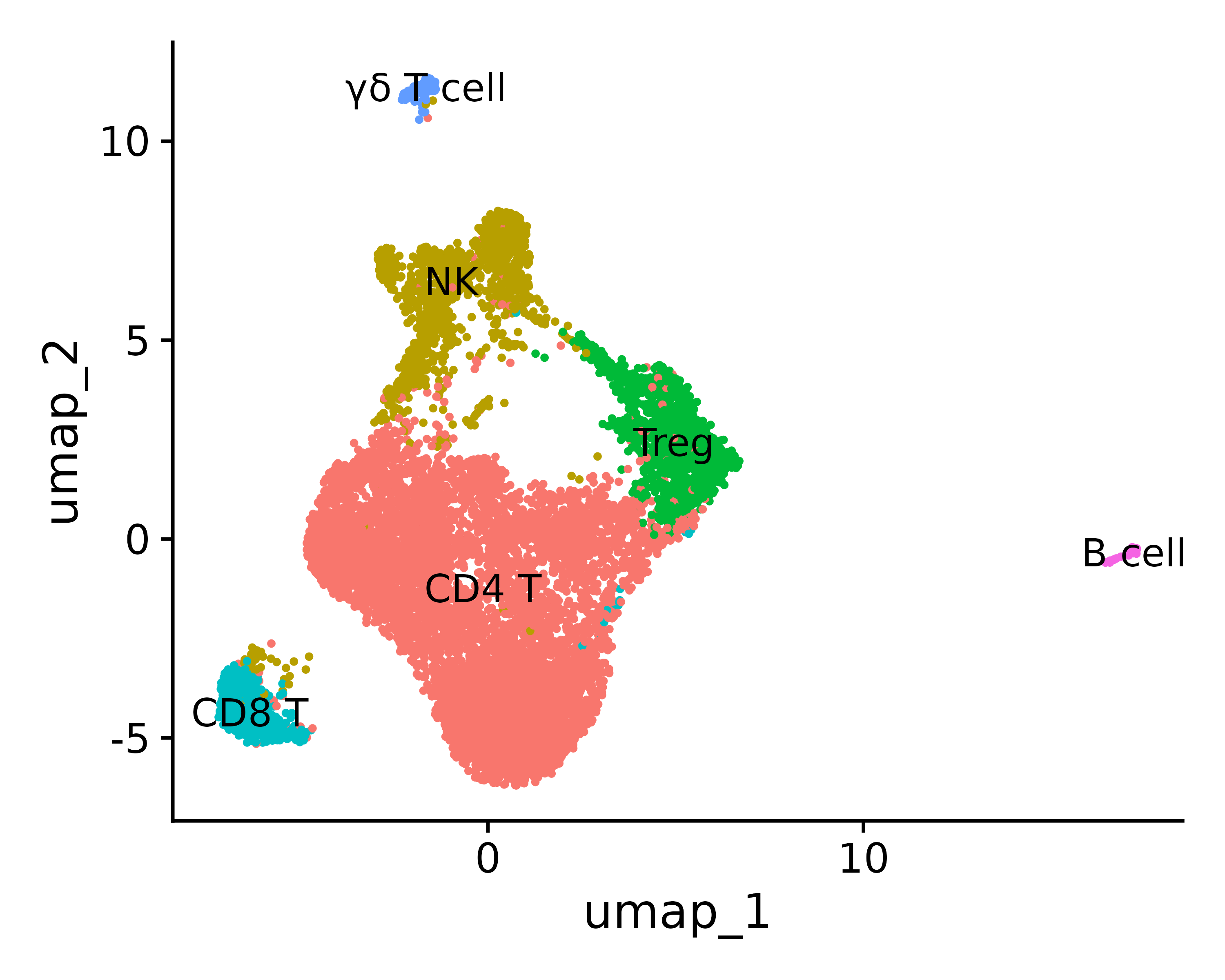
Referenz
KI mit KI entwickeln
Von der Idee bis zum Launch – beschleunigen Sie Ihre KI-Entwicklung mit kostenlosem KI-Co-Coding, sofort einsatzbereiter Umgebung und bestem GPU-Preis.