Command Palette
Search for a command to run...
Durch Die Verknüpfung Von Genexpressionsdaten Mit Bildern Der Zellmorphologie Haben Forscher Der Chinesischen Universität Hongkong Und Anderer Universitäten Ein Transkriptomgesteuertes Diffusionsmodell Entwickelt, Um Die Phänotypische Arzneimittelentwicklung Zu beschleunigen.
Die Zellmorphologie ist ein zentrales Forschungsgebiet der Einzelzellbiologie. Ihr Wert liegt in der systematischen Analyse der dynamischen Veränderungen der Zellmorphologie unter genetischen oder medikamentösen Einflüssen mittels Hochdurchsatz-Bildanalyse. Diese Forschung verbessert nicht nur die Genauigkeit von Vorhersagen zum Wirkmechanismus von Verbindungen (MOA) deutlich, sondern steigert auch die Genauigkeit von Bewertungen der Bioaktivität von Verbindungen. Letztlich liefert sie Datenunterstützung für wichtige Schritte der phänotypischen Arzneimittelforschung, wie z. B. das Screening von Wirkstoffkandidaten und die Verifizierung von Wirkmechanismen, und beschleunigt so den F&E-Prozess effektiv.
Die Beobachtung und Analyse von Veränderungen der Zellmorphologie nach genetischen oder medikamentösen Eingriffen ist jedoch keine einfache Aufgabe. Die Zahl der zu untersuchenden Verbindungen übersteigt Millionen, und auch die Zahl der editierbaren Gene liegt im Zehntausendenbereich. Die Verwendung herkömmlicher experimenteller Methoden zur individuellen Überprüfung jedes einzelnen Moleküls ist nicht nur äußerst ineffizient, sondern auch zeit- und kostenintensiv. Obwohl verschiedene computergestützte Methoden zur Vorhersage der Zellmorphologie vorgeschlagen und angewendet wurden, genügen ihre Genauigkeit und Genauigkeit den Anforderungen der praktischen Forschung noch immer nicht.
Speziell,Die Einschränkungen bestehender Methoden spiegeln sich hauptsächlich in zwei Punkten wider:Erstens ist die Leistungsfähigkeit fortgeschrittener Modelle wie IMPA (IMage Perturbation Autoencoder) stark von bekannten biologischen Kenntnissen oder spezifischen Datensätzen abhängig, was zu schwachen Generalisierungsmöglichkeiten und mangelnder breiter Anwendbarkeit führt. Zweitens werden Zellmorphologiedaten leicht durch experimentelle Störfaktoren wie Batch-Effekte und Well-Position-Effekte beeinflusst und weisen ein hohes Rauschen auf, was es schwierig macht, die wahren zellmorphologischen Eigenschaften effektiv zu erfassen, was wiederum die Stabilität und Zuverlässigkeit der Daten direkt beeinträchtigt und die Genauigkeit nachfolgender Analyseergebnisse einschränkt.
Um die oben genannten Herausforderungen zu bewältigen, haben Forscher der Chinesischen Universität Hongkong, der Mohamed bin Zayed University of Artificial Intelligence und anderer Institutionen ein skalierbares, transkriptomgesteuertes Diffusionsmodell namens MorphDiff vorgeschlagen.Dieses Modell ist speziell für die hochpräzise Simulation der Reaktion der Zellmorphologie auf Störungen konzipiert. Es basiert auf der Architektur des Latent Diffusion Model (LDM) und verwendet das L1000-Genexpressionsprofil als bedingten Input für das Denoising-Training.
Die Ergebnisse der Studie bestätigten, dassDer Hauptvorteil von MorphDiff ist seine Fähigkeit, Zellmorphologien unter „ungesehenen Störungsbedingungen“ genau zu generieren.Diese Fähigkeit bietet zwei entscheidende Vorteile: Erstens hilft sie Forschern, den weiten Bereich des phänotypischen Störungsscreenings effizient zu erkunden, wodurch die Abhängigkeit von groß angelegten Feldversuchen deutlich reduziert wird, was wiederum die Versuchskosten senkt und die Screening-Effizienz verbessert. Zweitens hilft sie bei der Aufklärung der Wirkmechanismen strukturell unterschiedlicher Arzneimittelmoleküle und liefert wichtige Unterstützung für die Validierung von Wirkstoffmechanismen. MorphDiff kann daher als leistungsstarkes Tool zur Beschleunigung der phänotypischen Arzneimittelentwicklung dienen.
Die Forschungsergebnisse wurden in Nature Communications unter dem Titel „Prediction of cellular morphology changes under perturbations with a transcriptome-guided diffusion model“ veröffentlicht.
Forschungshighlights:
* In dieser Studie werden Diffusionsmodelle erstmals auf innovative Weise zur Vorhersage der Zellmorphologie angewendet. Dies eröffnet neue Wege und bietet neue Werkzeuge für die phänotypische Arzneimittelentwicklung.
* Umfangreiche Benchmarktests belegen die Effektivität von MorphDiff, insbesondere bei der MOA-Abfrage, wo es eine mit der Ground-Truth-Morphologie vergleichbare Genauigkeit erreichen kann und die Basismethoden um 16,9% bzw. 8% übertrifft.

Papieradresse:
https://www.nature.com/articles/s41467-025-63478-z
Folgen Sie dem offiziellen Konto und antworten Sie mit „transcriptome-guided diffusion“, um das vollständige PDF zu erhalten
Weitere Artikel zu den Grenzen der KI:
https://hyper.ai/papers
Datensatz: Umfangreicher Multi-Omics-Datensatz zur Überprüfung seiner Wirksamkeit
Um die Wirksamkeit und Generalisierbarkeit des MorphDiff-Modells bei der Vorhersage der Zellmorphologie unter Störungsbedingungen systematisch zu überprüfen,Diese Studie konstruierte ein Multizelllinien- und Multiquellen-Datensatzsystem basierend auf den dualen Dimensionen „genetische Störung – Arzneimittelstörung“.Für jede Probe im Experiment wurden zwei Arten von Daten erhoben: ein L1000-Genexpressionsprofil und ein Zellmorphologiebild, die gepaarte Daten bildeten. Ersteres diente als „molekularer Merkmalseingang“, letzteres als „phänotypischer Merkmalseingang“. Diese Methode stellt die Korrelation zwischen dem Störungssignal auf genetischer Ebene und der phänotypischen Reaktion auf morphologischer Ebene sicher, die ausschließlich durch die Zielstörung gesteuert wird. Dadurch werden Störungen durch irrelevante Variablen wie Zelllinienunterschiede und Versuchschargen eliminiert.
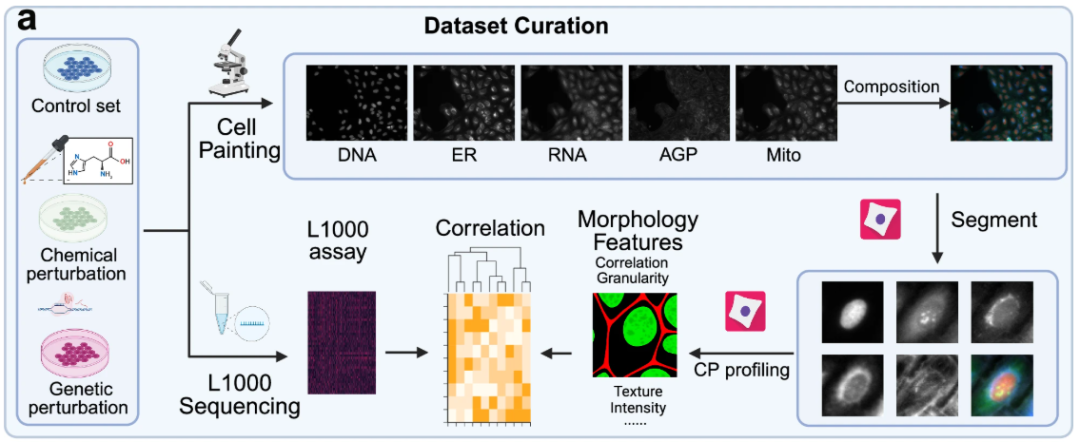
In Bezug auf Bilddatensätze zur Zellmorphologie,Für die Studie wurden drei umfangreiche Bilddatensätze zur Zellmorphologie für das Training, die Auswertung und die Analyse des Modells verwendet: ein Datensatz zu genetischen Störungen und zwei Datensätze zu Arzneimittelstörungen. Der Datensatz zu genetischen Störungen, abgeleitet aus dem JUMP-Datensatz auf Basis der U2OS-Zelllinie, enthält 130 genetische Störungen. Die Datensätze zu Arzneimittelstörungen, abgeleitet aus dem CDRP-Datensatz auf Basis der U2OS-Zelllinie und dem LINCS-Datensatz auf Basis der A549-Zelllinie, enthalten 1.028 bzw. 61 Arzneimittelstörungen.
Alle drei Zellmorphologie-Bilddatensätze wurden vorverarbeitet und segmentiert. CellProfiler 4.2.5 wurde verwendet, um die Zellplattenbilder in Einzelzellbilder zu segmentieren und so eine detailliertere Analyse zu ermöglichen.Die mit der Cell Painting-Technologie erhaltenen Zellmorphologiebilder enthalten 5 Kernkanäle.Das heißt, DNA (Zellkern), RNA (Nukleolus und Zytoplasma), ER (Endoplasmatisches Retikulum), AGP (Golgi-Apparat/Zellmembran/Aktinskelett) und Mito (Mitochondrien).
außerdem,Das Experiment plante außerdem den L1000-Datensatz „ohne entsprechende morphologische Bilder“.Dieser Datensatz wird hauptsächlich verwendet, um die Anwendung des Modells im Szenario „nur Genexpressionsdaten erhalten“ weiter zu untersuchen und kann die Grundlage für die anschließende Mechanismusüberprüfung, Arzneimittelscreening usw. legen, um „datengesteuerte“ Hypothesen bereitzustellen.
Modellarchitektur und Methoden: Verknüpfung von Genexpressionsdaten mit Zellmorphologiebildern
Das Hauptziel von MorphDiff besteht darin, durchgängig eine genaue Zuordnung von L1000-Genexpressionsprofilen zu Zellmorphologiebildern mithilfe eines transkriptomgesteuerten latenten Diffusionsmodell-Frameworks zu erreichen.Einfach ausgedrückt geht es darum, ein Modell zu entwerfen und zu trainieren, das wie eine „Brücke“ funktioniert: Geben Sie die L1000-Genexpressionsdaten ein, die einer bestimmten „Störung“ entsprechen, und geben Sie dann das morphologische Bild oder die Störungsmorphologie der Zelle unter dieser Störung aus.
Der Kern des MorphDiff-Modells besteht aus zwei Hauptmodulen:Morphology Variational Autoencoder (MVAE) und Latent Diffusion Model (LDM), wie in Abbildung b unten dargestellt.
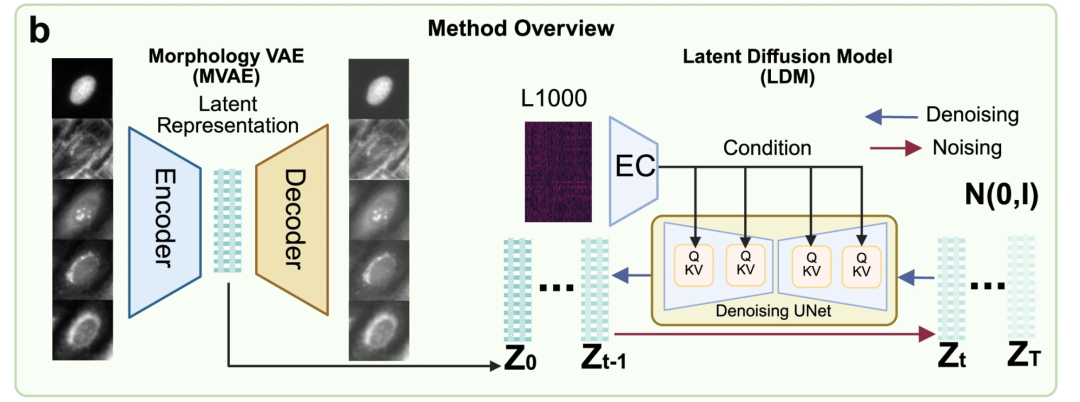
Unter ihnen ist MVAE die „Bildkomprimierungs-Engine“ des Modells, die für die Konvertierung mehrkanaliger, hochauflösender Zellmorphologiebilder in niedrigdimensionale, interpretierbare Potenzialdarstellungen verantwortlich ist.Dieser Ansatz behebt den hohen Rechenaufwand und das instabile Training, die mit dem direkten Training von Diffusionsmodellen auf hochdimensionalen Bildern verbunden sind. Strukturell besteht das MVAE aus zwei Teilen: einem Encoder und einem Decoder. Der Encoder komprimiert komplexe 5-Kanal-Morphologiebilder in eine einfachere niedrigdimensionale Darstellung; der Decoder stellt das ursprüngliche Morphologiebild aus dieser niedrigdimensionalen Darstellung wieder her.
LDM ist hauptsächlich für die Verknüpfung von „Genexpression“ und „komprimierten morphologischen Merkmalen“ verantwortlich und hilft dem Modell, die Beziehung „von Genen zu morphologischen Merkmalen“ zu vervollständigen. LDM umfasst einen Entrauschungsprozess und einen Entrauschungsprozess. Der Entrauschungsprozess fügt den komprimierten morphologischen Merkmalen schrittweise Gaußsches Rauschen hinzu, bis diese vollständig zufällig sind. Der Entrauschungsprozess ermöglicht es dem Modell, das zufällige Rauschen schrittweise auf die ursprünglichen morphologischen Merkmale zurückzuführen, vorausgesetzt die Expression der L1000-Gene ist bekannt. Das Modell verwendet eine U-Net-Netzwerkarchitektur und integriert einen Aufmerksamkeitsmechanismus, um wichtige genetische und morphologische Informationen genauer zu verknüpfen.
Abbildung c unten zeigt zwei Anwendungen des vortrainierten MorphDiff-Modells: G2I und I2I. Ersteres entrauscht das entsprechende Zellmorphologiebild anhand einer zufälligen Rauschverteilung, konditioniert durch die L1000-Genexpression, um das entsprechende Zellmorphologiebild zu erzeugen. Letzteres, konditioniert durch die L1000-Genexpression unter einer bestimmten Störung, transformiert das Morphologiebild von der Kontrollzellmorphologie in das vorhergesagte Bild der gestörten Morphologie und ermöglicht so die Vorhersage von „normaler bis gestörter Morphologie“.
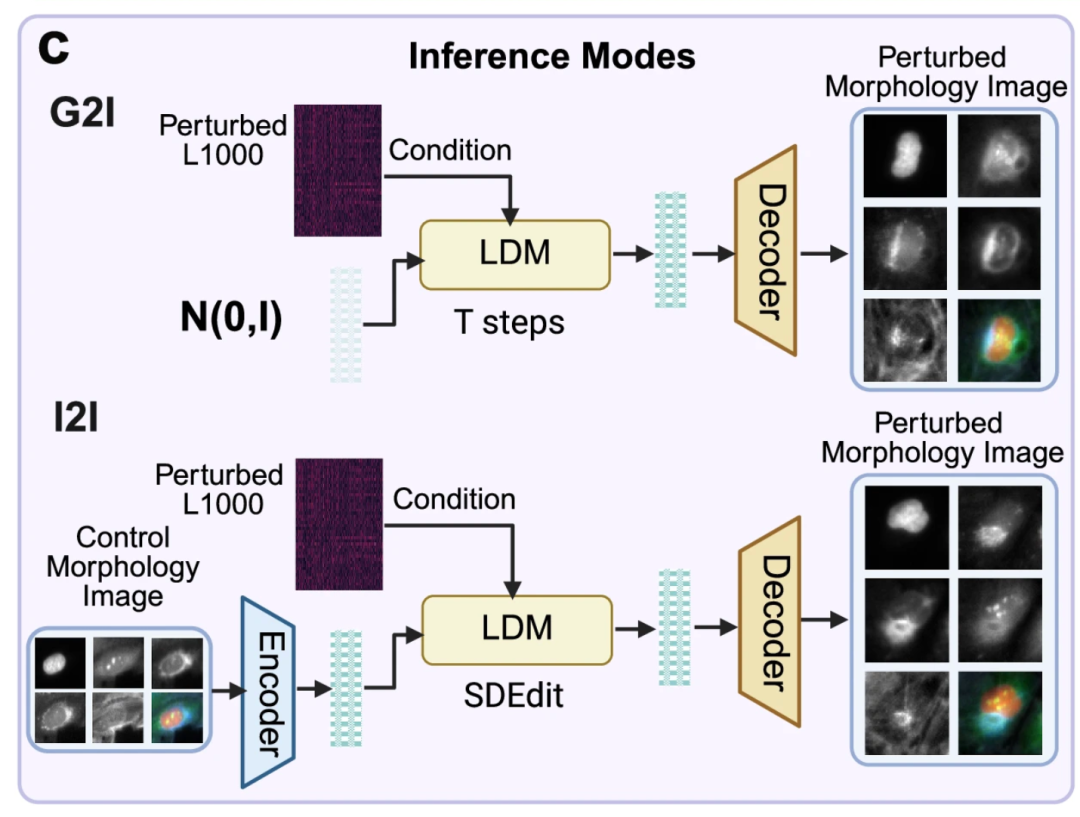
Abbildung d unten zeigt den Wert des MorphDiff-Modells in praktischen Anwendungen.Erstens ist das Modell in der Lage, zellmorphologische Veränderungen vorherzusagen, die durch „unsichtbare Störungen“ verursacht werden, die während des Trainings nicht aufgetreten sind.Dies ermöglicht es Forschern, den Zustand von Zellveränderungen unter der Einwirkung neuer Medikamente am Computer zu simulieren, ohne physikalische Experimente durchführen zu müssen. So können sie schnell und kostengünstig mehr Möglichkeiten erkunden.Das Modell-Framework kombiniert Tools wie CellProfiler und DeepProfiler.Es kann dabei helfen, den MOA von Medikamenten zu identifizieren und so die Entwicklung phänotypischer Medikamente zu fördern.
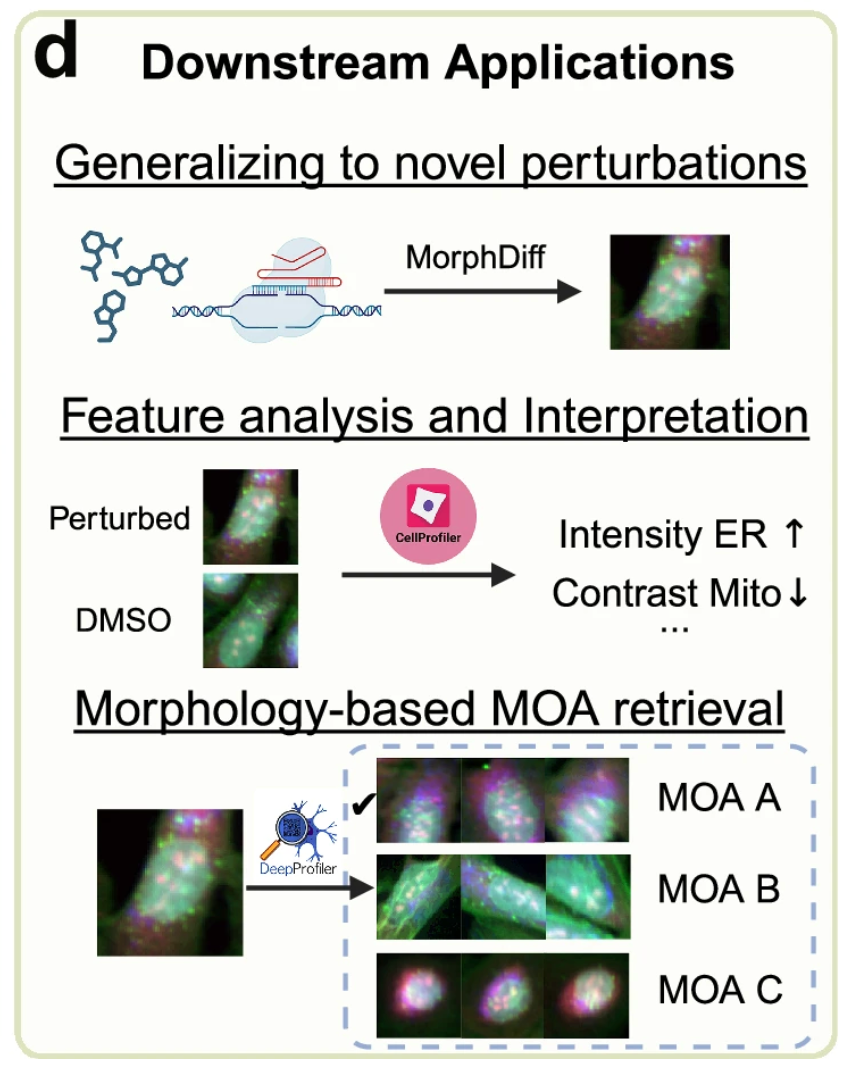
Den Forschern zufolge ist MorphDiff derzeit das einzige Tool, das die Generierung morphologischer Bilder aus der Genexpression und die Konvertierung von ungestörter Morphologie in gestörte Morphologie unterstützt.
Experimentelle Ergebnisse: Die Leistung übertrifft IMPA und beschleunigt die phänotypische Arzneimittelentwicklung
Um die Wirksamkeit des MorphDiff-Modells zu überprüfen, haben die Forscher eine Reihe von Experimenten für verschiedene Zwecke entwickelt.Durch experimentelle Vergleiche mit fortschrittlichen Tools werden die Wirksamkeit und Praktikabilität von MorphDiff systematisch überprüft.
Erstens bestätigten die Experimente die Vorhersagen zur genetischen Störung.Die Forscher führten Benchmarktests mit dem JUMP OOD-Datensatz durch und verglichen sie mit mehreren Basismethoden, darunter MorphNet, DMIT (Disentanglement for Multi-mapping Image-to-Image Translation), DRIT++ (Disentangled Representation for Image-to-Image Translation), StarGANv1, IMPA, VQGAN (Vector Quantized Generative Adversarial Network) und MDTv2 (Masked Diffusion Transformers).
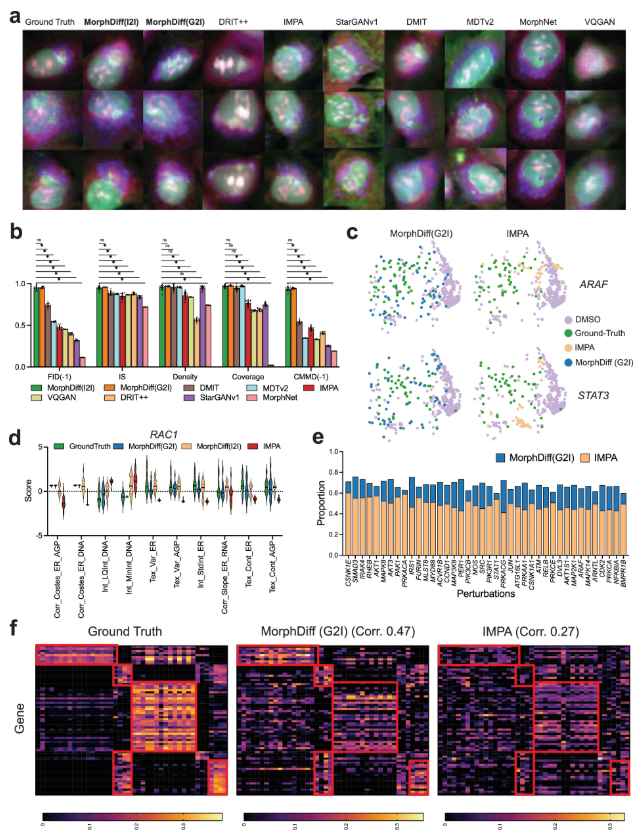
Die Ergebnisse zeigen, dassDie von den beiden MorphDiff-Modi generierten Ergebnisse liegen hinsichtlich visueller Qualität und struktureller Morphologie näher an der tatsächlichen Basislinie.In Bezug auf quantitative Metriken, einschließlich FID, IS (Inception Score), CMMD, Dichte und Abdeckung, übertrafen beide MorphDiff-Modi die Basismethoden in Bezug auf Generalisierung, Genauigkeit und Diversität und erzielten eine höhere Ausgabequalität. Bei der Vorhersage von Zellmorphologieänderungen sind die MorphDiff-Ausgaben (G2I) vielfältiger und näher an der tatsächlichen Basislinie; während die von MorphDiff (I2I) generierten Merkmale eine höhere Überlappung mit den tatsächlichen Basislinienmerkmalen aufweisen, was darauf hindeutet, dass die Vorhersagegenauigkeit weitgehend mit der tatsächlichen Störungsmorphologie übereinstimmt.
Anschließend wurden Experimente durchgeführt, um die Vorhersage der Arzneimittelstörung zu überprüfen. Die Forscher führten zunächst ein Benchmarking aller Methoden anhand des CDRP OOD-Datensatzes durch.MorphDiff erzielt im Vergleich zu anderen Basismethoden in den meisten Metriken eine beeindruckende Leistung und demonstriert damit seine leistungsfähigeren und stabileren umfassenden Generierungsfunktionen.Anschließend führten die Forscher eine strengere Bewertung des Modells anhand des LINCS-Datensatzes durch und untersuchten die Reaktion der Zellmorphologie auf kleine Molekülverbindungen weiter, indem sie die generierten Ergebnisse mit den Funktionen von CellProfiler verglichen.
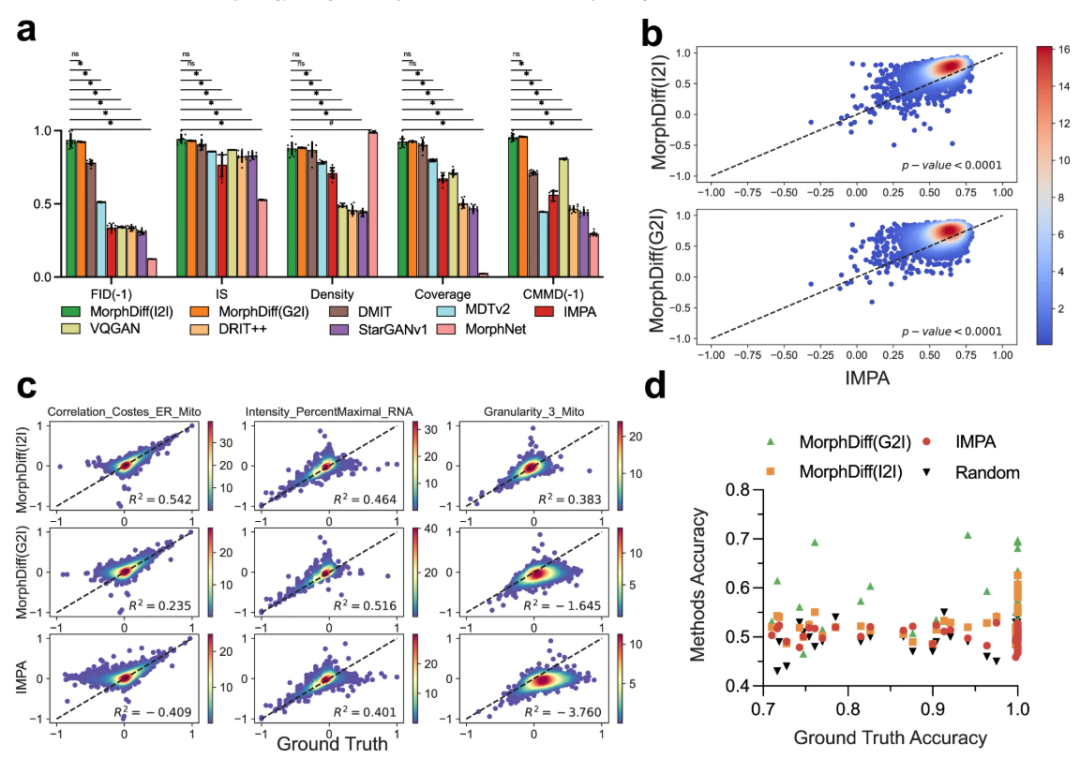
Die Ergebnisse zeigen, dass beide MorphDiff-Modi die Basislinie deutlich übertreffen.Unter G2I erzielten 87,61 TP3T-Proben einen Wert über 0,5 und 16,21 über 0,8. Unter I2I erzielten 891 TP3T-Proben einen Wert über 0,5 und 27,21 über 0,8. Im Vergleich dazu erzielten 78,31 TP3T-Proben von IMPA einen Wert über 0,5, aber keine über 0,8. Die gleiche Analyse des CDRP OOD-Datensatzes und des LINCS Leave-One-Out-Datensatzes ergab, dass beide MorphDiff-Modelle die Basismethode mit p-Werten unter 0,0001 übertrafen, was die Generalisierbarkeit der Methode belegt.
In der DeepProfiler-EinbettungsanalyseMorphDiff (G2I) kann störungsspezifische zellmorphologische Muster am zuverlässigsten und genauesten erfassen.Auf pharmazeutischer EbeneDie vom G2I-Muster erzeugte Ausgabe weist eine höhere Störungsspezifität auf als die von I2I. Diese Demonstrationen verdeutlichen das Potenzial von MorphDiff im Arzneimittelscreening.
Schließlich überprüfte das Experiment auch die Fähigkeiten von MorphDiff in der Arzneimittelentwicklung. Die Forscher wählten den CDRP Target_MOA-Datensatz aus, um die beiden Anwendungsmodi und IMPA von MorphDiff zu vergleichen.
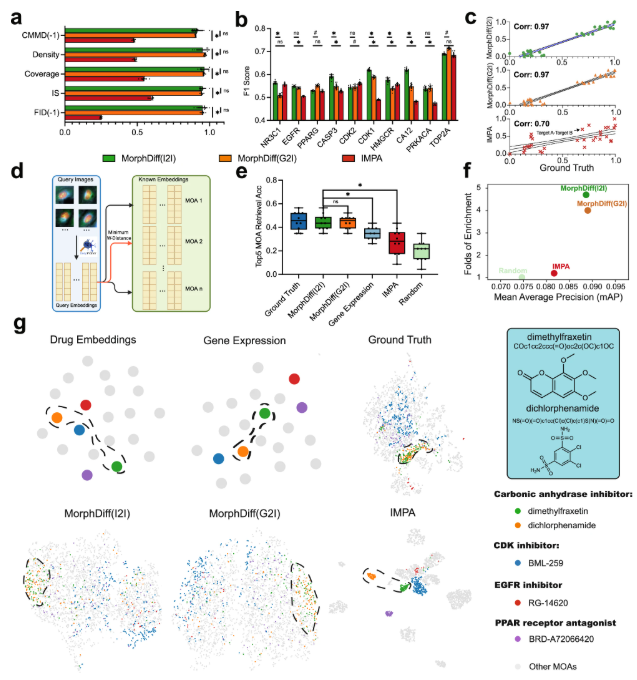
Die Ergebnisse zeigen, dass die von MorphDiff generierte Störungsmorphologie in hohem Maße mit der wahren Basislinie übereinstimmt.Dies demonstrierte seine Fähigkeit, zielbezogene Diversität zu erfassen und die komplexe Beziehung zwischen Arzneimittelstörungen und Morphologie effektiv zu erlernen. Bei der MOA-Abrufaufgabe war die durchschnittliche Genauigkeit der von MorphDiff generierten Ergebnisse 16,91 TP3T höher als bei IMPA-basierter und 81 TP3T höher als bei genexpressionbasierter Abfrage. Darüber hinaus zeigten Experimente, dass MorphDiff erkennen kann, dass die Zellmorphologie komplementäre Informationen enthält, und Arzneimittel mit demselben MOA, aber unterschiedlichen Strukturen identifizieren kann, was die phänotypische Arzneimittelentwicklung beschleunigen kann.
Siliziumbasierte Simulationen werden zum Mainstream und beschleunigen die Arzneimittelentwicklung
Die phänotypische Wirkstoffforschung unterscheidet sich von der zielbasierten Wirkstoffforschung dadurch, dass sie Medikamente erforscht und entwickelt, indem ihre Auswirkungen auf biologische Systeme oder zelluläre Phänotypen beobachtet werden. Dieser Ansatz bietet erhebliche Vorteile bei der Entdeckung neuer Wirkmechanismen und Zielstrukturen sowie bei der Behandlung komplexer Erkrankungen. Zahlreiche Labore und Forschungseinrichtungen verfolgen dieses Thema aktiv und integrieren Computertechnologie mit Biomedizin, um ein neues Kapitel in der phänotypischen Wirkstoffforschung aufzuschlagen.
Wie in dem Artikel erwähnt, handelt es sich beim IMPA-Modell um ein tiefes generatives Modell, das von einem Team der Technischen Universität München in Deutschland und der Universität Oxford in Großbritannien vorgeschlagen wurde.Mithilfe der Stilübertragungsmethode werden Zellbilder in „Stil“ (Störung/Batch-Darstellung) und „Inhalt“ (Zelldarstellung) zerlegt, wodurch die Reaktion der Zelle auf Störungen vorhergesagt und Batch-Effekte entfernt werden können.Der Artikel mit dem Titel „Vorhersage zellmorphologischer Reaktionen auf Störungen mithilfe generativer Modellierung“ wurde auch in Nature Communications veröffentlicht.
Darüber hinaus heißt es in einem vom Team der University of Michigan veröffentlichten Artikel mit dem Titel „MorphNet Predicts Cell Morphology from Single-Cell Gene Expression“:schlug eine rechnergestützte Methode namens MorphNet vor, die morphologische Bilder von Zellen auf der Grundlage ihrer Genexpressionsprofile zeichnen kann.Die Methode verwendet gepaarte morphologische und molekulare Daten, um ein neuronales Netzwerk zu trainieren, das die Morphologie des Zellkerns oder der gesamten Zelle basierend auf der Genexpression vorhersagt.
Zusammenfassend lässt sich sagen, dass die Förderung der phänotypischen Arzneimittelentwicklung und der biologischen Forschung durch die Beobachtung und Analyse von Veränderungen des Zellzustands unter genetischen oder medikamentösen Störungen zu einem wichtigen Thema geworden ist. MorphDiff weist zwar noch viele Mängel auf, wie beispielsweise die Herausforderung, angesichts einer großen Anzahl neuer Störungen außerhalb der Trainingsdaten ungesehene Störungen vorherzusagen, doch ist klar, dass MorphDiff auf der Grundlage seiner Vorgänger ständig verbessert wird und diese in Bezug auf Praktikabilität, Generalisierung, Benutzerfreundlichkeit und Skalierbarkeit übertrifft.
Erhalten Sie mit einem Klick hochwertige Papiere und ausführliche Interpretationsartikel im Bereich AI4S von 2023 bis 2024 ⬇️









