Command Palette
Search for a command to run...
Neudefinition Der Klassifizierung Von Protein-Sprachmodellen Basierend Auf Der Beziehung Zwischen Struktur/Sequenz/Funktion: Dr. Li Mingchen Erklärt Protein-Sprachmodelle Im Detail

Die dritte „AI for Bioengineering Summer School“ der Shanghai Jiao Tong University wurde vom 8. bis 10. August 2025 offiziell eröffnet.Diese Sommerschule brachte mehr als 200 junge Talente, wissenschaftliche Forscher und Industrievertreter von über 70 Universitäten, mehr als 10 wissenschaftlichen Forschungseinrichtungen und mehr als 10 branchenführenden Unternehmen aus der ganzen Welt zusammen und konzentrierte sich auf die integrierte Entwicklung von künstlicher Intelligenz (KI) und Bioengineering.
Unter anderem stellte Li Mingchen, Postdoktorand in der Forschungsgruppe Hong Liang am Institut für Naturwissenschaften der Jiao Tong-Universität Shanghai, im Kursabschnitt „Grenzen der KI-Algorithmen“ allen die neuesten Errungenschaften von Proteinsprachenmodellen bei der Funktionsvorhersage, Sequenzgenerierung, Strukturvorhersage usw. zum Thema „Grundmodell von Proteinen und Genomen“ sowie die damit verbundenen Forschungsfortschritte bei Expansionsgesetzen und Genommodellen vor.

HyperAI hat die wunderbaren Ausführungen von Dr. Li Mingchen zusammengestellt und zusammengefasst, ohne die ursprüngliche Absicht zu verletzen. Im Folgenden finden Sie die Essenz der Rede.
Eine neue Klassifizierung von Protein-Sprachmodellen: Die Beziehung zwischen Proteinstruktur, -sequenz und -funktion
Proteine haben ein breites Anwendungsspektrum in Bereichen wie Chemieingenieurwesen, Landwirtschaft, Lebensmittel, Kosmetik, Medizin und Testverfahren. Ihr Marktwert übersteigt Billionen von Dollar. Vereinfacht ausgedrückt handelt es sich bei der Modellierung der Proteinsprache um ein Problem der Wahrscheinlichkeitsverteilung. Dies entspricht der Bestimmung der Wahrscheinlichkeit des Vorkommens einer Aminosäuresequenz in der Natur und der entsprechenden Stichprobenziehung. Durch Vortraining mit riesigen Datenmengen kann das Modell die in der Natur vorkommende Wahrscheinlichkeitsverteilung effektiv abbilden.
Das Proteinsprachenmodell hat drei Kernfunktionen:
* Der Lernprozess der Darstellung von Proteinsequenzen als hochdimensionale Vektoren
* Bestimmen Sie die Rationalität der Aminosäuresequenz
* Generieren Sie neue Proteinsequenzen
Viele Forschungsarbeiten kategorisieren Proteinsprachenmodelle nach ihrer Transformer-Architektur und beschreiben sie direkt als Transformer-Encoder- oder Transformer-Decoder-basiert. Diese Klassifizierung ist für Biologen schwer verständlich und führt oft zu Verwirrung. Daher stelle ich eine neue Klassifizierungsmethode vor:Klassifizierung basierend auf der Beziehung zwischen Proteinstruktur, -sequenz und -funktion.
Die Sequenz eines Proteins ist seine Aminosäureabfolge. Ist die Aminosäuresequenz bekannt, kann das Protein im Labor oder in der Fabrik synthetisiert und in der Praxis eingesetzt werden. Ebenso entscheidend ist die Struktur eines Proteins. Seine Funktion beruht auf seiner spezifischen Struktur im dreidimensionalen Raum, die seine Funktion auf mikroskopischer Ebene ermöglicht.
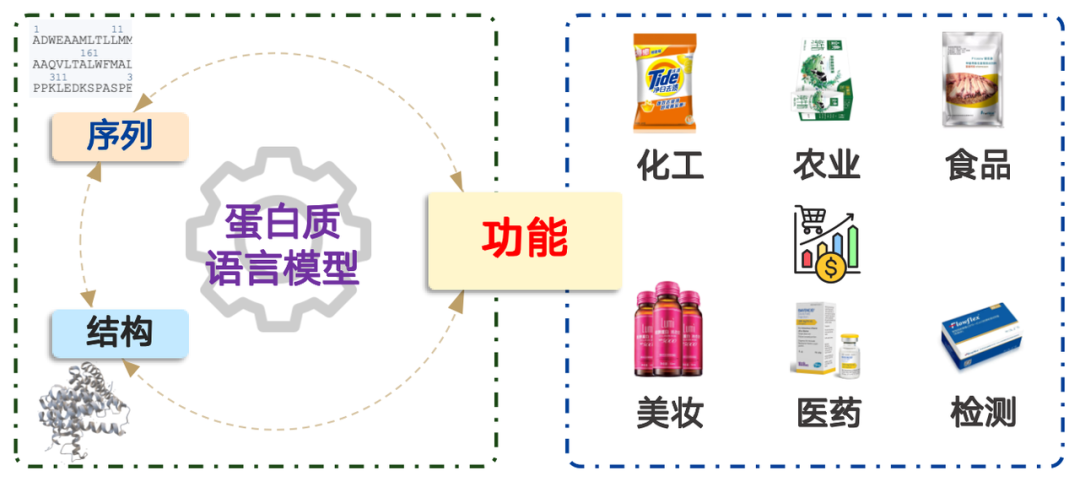
Basierend auf dieser Idee können Proteinsprachenmodelle in die folgenden vier Kategorien unterteilt werden:
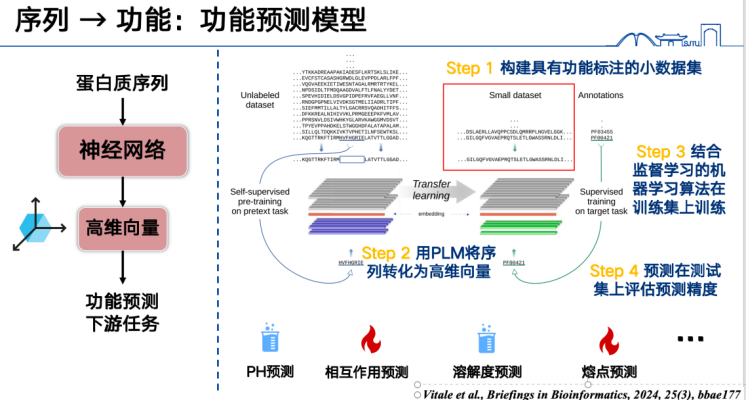
1. Ablauf → Funktion:Vorhersage der Funktion einer gegebenen Aminosäuresequenz, d. h. Funktionales Vorhersagemodell.
2. Funktion → Ablauf:Entwerfen Sie die entsprechende Aminosäuresequenz entsprechend der gegebenen Funktion, einschließlich Generative ModelleUnd Bergbaumodell.
3. Reihenfolge → Struktur:Die Vorhersage seiner Struktur auf der Grundlage seiner Aminosäuresequenz wird üblicherweise als "Strukturelles Vorhersagemodell",Zu diesem Modelltyp gehört das mit dem Nobelpreis ausgezeichnete AlphaFold.
4. Struktur → Reihenfolge:Das Entwerfen einer entsprechenden Sequenz basierend auf einer gegebenen Proteinstruktur wird üblicherweise als „Umgekehrtes Faltmodell“.
Anwendungsszenarien und technische Pfade: Analyse von vier Mainstream-Modellen
"Sequenz → Funktion"
Der einfachste Weg, „Sequenz → Funktion“ zu verstehen, ist überwachtes Lernen.
Das grundlegendste Modell zur Funktionsvorhersage besteht zunächst darin, Proteinsequenzen als Vektoren darzustellen und diese dann anhand eines bestimmten Datensatzes zu trainieren. Um beispielsweise Proteinschmelzpunkte vorherzusagen, müssen wir zunächst eine große Menge an Proteinschmelzpunktmarkierungen sammeln, alle Proteinsequenzen im Trainingssatz in hochdimensionale Vektoren umwandeln und diese mithilfe überwachter Lernmethoden trainieren. Anschließend können wir Inferenzen auf Sequenzen im Test- oder Vorhersagesatz anwenden, um die Funktion vorherzusagen. Dieser Ansatz eignet sich für ein breites Aufgabenspektrum und ist derzeit ein aktuelles Forschungsthema, das zudem relativ einfach Ergebnisse liefert.
Zweitens kann das Proteinsprachenmodell auch Mutationsfunktionen vorhersagen.Die Kernidee besteht darin, einige Änderungen an bestimmten Aminosäuren in der Proteinsequenz vorzunehmen und dann mithilfe des Proteinsprachenmodells zu bestimmen, ob die Änderung „sinnvoll“ ist.
Mit „vernünftig“ ist hier nicht die logische Übereinstimmung im wirklichen Leben gemeint, sondern vielmehr die Frage, ob die Aminosäureänderung der Wahrscheinlichkeitsverteilung natürlicher Proteinsequenzen entspricht. Diese Wahrscheinlichkeitsverteilung basiert auf einer großen Zahl realer Aminosäuresequenzstatistiken, und diese Aminosäureverteilungen sind selbst das Produkt von zig Millionen Jahren Evolution.
Das Proteinsprachenmodell lernt diese evolutionären Gesetze während des Trainings und kann daher bestimmen, ob eine Mutation diesen Gesetzen entspricht oder von ihnen abweicht. Mathematisch lässt sich diese Bestimmung in das Verhältnis der Wahrscheinlichkeiten der beiden Sequenzen vor und nach der Mutation umwandeln. Zur Vereinfachung der Berechnung wird dieses Verhältnis oft logarithmiert und so in eine Subtraktionsform umgewandelt.
Das von Sprachmodellen verwendete Likelihood-Verhältnis zwischen Mutanten und Wildtyp-Proteinen kann die Stärke der Wirkung einer Mutation abschätzen. Diese Idee wurde erstmals 2018 in einem Nature Methods-Artikel zur Einführung des DeepSequence-Modells demonstriert, das damals jedoch noch relativ klein war. Anschließend, im Jahr 2021, zeigte das ESM-1v-Modell weiter, dass Protein-Sprachmodelle Mutationseffekte ebenfalls mithilfe von Likelihood-Verhältnissen effektiv vorhersagen können.
Um die Genauigkeit des Vorhersagemodells für die Proteinmutationsfunktion zu bewerten, ist ein Benchmark erforderlich.
Benchmarks sind kleine Datensätze, die zur Messung der Genauigkeit gesammelt werden. Der am häufigsten verwendete Benchmark ist beispielsweise ProteinGym, das gemeinsam von der Harvard Medical School und der Universität Oxford entwickelt wurde. Es enthält Daten zu 217 mutierten Proteinen und Millionen von Mutationssequenzen. Forscher weisen jeder dieser Mutationssequenzen mithilfe eines Proteinsprachenmodells Werte zu und vergleichen anschließend die vom Modell prognostizierten Werte mit den tatsächlichen Werten. Eine höhere Korrelation weist auf eine bessere Modellleistung hin.
ProteinGym ist jedoch ein Benchmark mit hohem Durchsatz und geringer Präzision.Obwohl es durch die experimentellen Bedingungen eingeschränkt ist, kann es in großem Maßstab getestet werden, die Genauigkeit kann jedoch begrenzt sein. Die Wiederholung eines Experiments kann zu Fehlern bei der Korrelation zwischen den Ergebnissen und den Originaldaten führen, wodurch die Auswertungsergebnisse die Leistung des Modells in realen Anwendungen ungenau widerspiegeln.
Um dieses Problem zu lösen,Wir haben einen Benchmark mit geringem Durchsatz und hoher Präzision für kleine Stichproben wie VenusMutHub entwickelt.Obwohl die Datenmenge nicht groß ist, ist jedes Datenelement relativ genau und die Ergebnisse wiederholter Experimente sind nahezu konsistent, was den realen Anwendungsszenarien näher kommt.
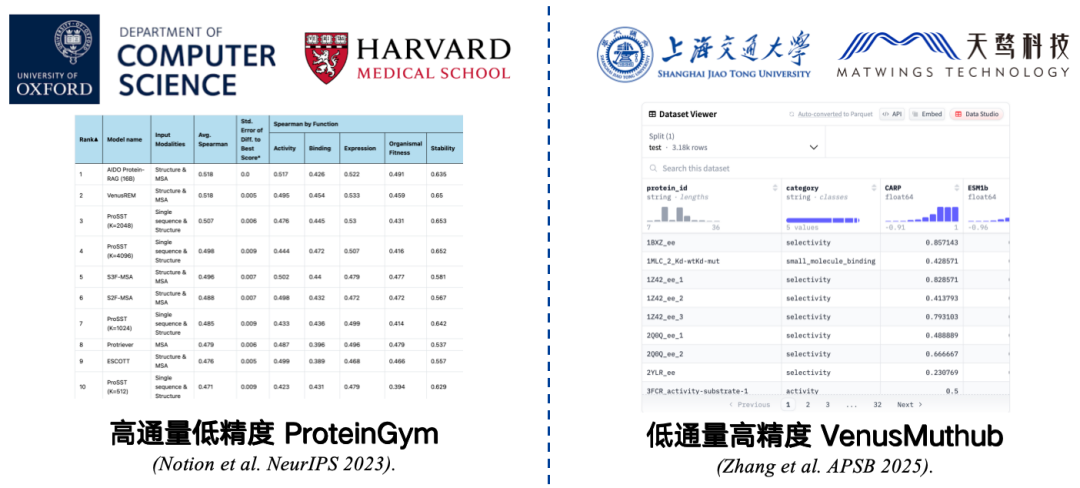
* Papieradresse:Zhang L, Pang H, Zhang C, et al. VenusMutHub: Eine systematische Bewertung von Prädiktoren für Proteinmutationseffekte anhand von experimentellen Daten im kleinen Maßstab[J]. Acta Pharmaceutica Sinica B, 2025, 15(5): 2454-2467.
Darüber hinaus kann die Einführung von Strukturen die Genauigkeit der Mutationsvorhersage von Proteinsprachenmodellen verbessern. Letztes Jahr veröffentlichte unser Team bei NeurIPS eine Arbeit über ein Proteinsprachenmodell, das ProSST-Modell. Dieses Modell verwendet sowohl Aminosäuresequenzen als auch strukturierte Sequenzen für multimodales Vortraining. ProSST belegte den ersten Platz im ProteinGym Benchmark, dem größten Benchmark zur Vorhersage von Mutationen ohne Schüsse.
* Papieradresse:Li M, Tan Y, Ma X, et al. ProSST: Proteinsprachenmodellierung mit quantisierter Struktur und entwirrter Aufmerksamkeit[C]. Fortschritte in der neuronalen Informationsverarbeitung, 2024, 37: 35700-35726.
Bei Experimenten oder Designs stößt man häufig auf folgende Fragen: „Welches Modell soll ich verwenden?“ „Wie soll ich als Benutzer auswählen?“
In einer in diesem Jahr veröffentlichten StudieUnser Team stellte fest, dass die Perplexität des Proteinsprachenmodells für die Zielsequenz in etwa dessen Genauigkeit bei der Mutationsvorhersage widerspiegeln kann.Der Vorteil besteht darin, dass eine Leistungsschätzung möglich ist, ohne dass Daten zu Zielproteinmutationen erforderlich sind. Je geringer die Perplexität, desto besser versteht das Modell die Sequenz, was oft bedeutet, dass seine Mutationsvorhersagen für diese Sequenz genauer sind.
Basierend auf dieser Idee haben wir ein Ensemble-Modell namens VenusEEM entwickelt. Es gewichtet Modelle basierend auf ihrer Perplexität oder wählt direkt das Modell mit der geringsten Perplexität aus. Dies verbessert die Genauigkeit der Mutationsvorhersage auf ein hohes Niveau. Unabhängig von der Strategie bleibt der endgültige Vorhersagewert relativ stabil, wodurch signifikante Leistungseinbußen durch falsche Modellauswahl vermieden werden.
* Papieradresse:Yu Y, Jiang F, Zhong B, et al. Entropiegesteuerte Zero-Shot-Deep-Learning-Modellauswahl für virale Proteine[J]. Physical Review Research, 2025, 7(1): 013229.
Schließlich entwickelte unser Team im Forschungsschwerpunkt „Sequenz zu Funktion“ im vergangenen Jahr zusätzlich zu den zuvor erwähnten Modellen auch ein neuartiges iteratives High-Site-Mutations-Designmodell namens PRIME. Konkret trainierten wir zunächst ein umfangreiches Proteinsprachenmodell anhand von 98 Millionen Proteinsequenzen. Für die Vorhersage von High-Site-Mutationen erhielten wir zunächst Low-Site-Mutationsdaten, gaben diese in das Proteinsprachenmodell ein und kodierten sie in einen Funktionsvektor. Basierend auf diesem Funktionsvektor trainierten wir dann ein Regressionsmodell zur Vorhersage von High-Site-Mutationen.Durch diese iterative Reaktion kann in nur 2–3 Versuchsrunden ein hervorragendes Proteinprodukt entwickelt werden.
* Papieradresse:Jiang F, Li M, Dong J, et al. Ein allgemeines temperaturgesteuertes Sprachmodell zur Entwicklung von Proteinen mit verbesserter Stabilität und Aktivität[J]. Science Advances, 2024, 10(48): eadr2641.

「Funktion→Sequenz」
Was wir zuvor besprochen haben, ist der Übergang von der Sequenz zur Funktion. Überlegen wir, ob wir die Sequenz umgekehrt aus der Funktion ableiten können.
Zwischen Folgen und Funktionen besteht ein Vorwärts- und ein Rückwärtsproblem. Während es beim Vorwärtsproblem darum geht, eine endgültige Antwort zu finden, geht es beim Rückwärtsproblem darum, innerhalb eines großen realisierbaren Raums nach einer lösbaren Lösung zu suchen. Das Generieren von Folgen aus Funktionen ist genau dieses Rückwärtsproblem. Der Grund dafür ist, dass Folgen typischerweise nur einer oder wenigen Funktionen entsprechen, eine einzelne Funktion jedoch durch eine Vielzahl völlig unterschiedlicher Folgen implementiert werden kann. Darüber hinaus gibt es keinen zuverlässigen Benchmark für das Rückwärtsproblem. Wenn ein Modell Folgen aus einer gegebenen Funktion generiert, kann dessen Genauigkeit in der Regel nur experimentell getestet werden.

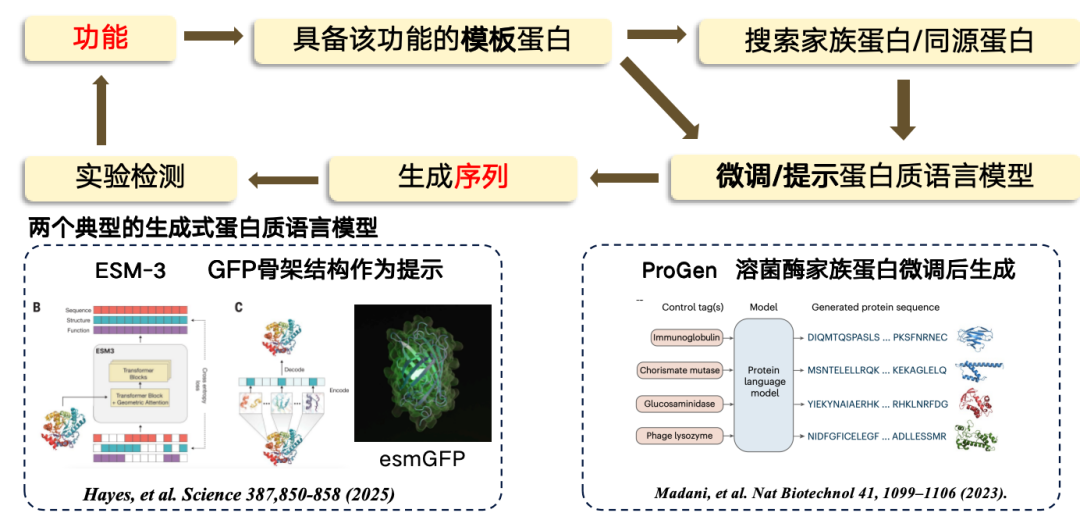
In der aktuellen StudieDer Entwurf von der Funktion zur Sequenz erfolgt meist nach einem Template-basierten Ansatz. Wenn bekannt ist, dass ein Template-Protein eine bestimmte Funktion hat, kann es als Grundlage für die Suche oder Generierung einer neuen Region verwendet werden.Der Prozess besteht darin, zunächst von der Funktion zur Vorlagensequenz zu gelangen, dann einige Familienproteine/homologe Proteine aus dem Vorlagenprotein zu suchen, dann das Proteinsprachenmodell zu verfeinern, das verfeinerte Sprachmodell zum Generieren neuer Sequenzbereiche zu verwenden und schließlich experimentelle Tests durchzuführen.
Zu den derzeit repräsentativsten generativen Proteinsprachenmodellen gehören:
* ESM-3, hergestellt unter Verwendung des grün fluoreszierenden Proteins (GFP) als Vorlage, das resultierende Protein ist jedoch weniger funktional.
* ProGen ist ein rein autoregressives Sprachmodell, ähnlich wie ChatGPT, das auf der Grundlage funktionaler Hinweise generiert werden kann. Es wird durch Feinabstimmung der Proteinarchitektur von Lysozym generiert.
Neben der direkten Generierung neuer Proteinsequenzen,Sie können auch direkt in der riesigen Menge vorhandener Proteinsequenzen suchen.Das Template-Protein wird in einem hochdimensionalen Raum kodiert, und der Abstand zwischen den Vektoren bestimmt, ob die beiden Proteine die gleiche Funktion haben. Abschließend werden die Ergebnisse aus einer Datenbank abgerufen. Das Prinzip dieses Ansatzes besteht darin, dass der Abstand zwischen den Kodierungen oder Vektoren zweier Proteine in einem hochdimensionalen Raum grob widerspiegelt, ob die beiden Proteine ähnliche Funktionen haben.
Die folgende Abbildung zeigt zwei typische Beispiele für das Mining von Protein-Sprachmodellen. Das erste ist ESM-Ezy, entwickelt von der Westlake University. Es nutzt das ESM-1b-Modell, um Vektorsuchen durchzuführen und mehrere Ausdrücke für die Auffüllung zu minen. Das zweite ist das groß angelegte Modell VenusMine, das hocheffiziente PET-Hydrolasen mint.
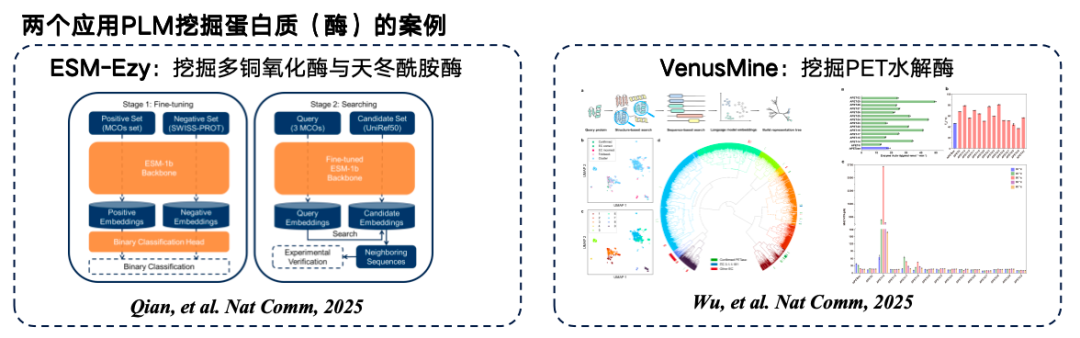
* Papieradresse:Wu B, Zhong B, Zheng L, et al. Nutzung eines Proteinsprachenmodells für die strukturbasierte Entdeckung hocheffizienter und robuster PET-Hydrolasen[J]. Nature Communications, 2025, 16(1): 6211.
Zusätzlich zu „Funktion → Sequenz“ können Sie zwischen Funktion und Sequenz auch „Vermittler“ hinzufügen:
* Wenn die Struktur als Vermittler verwendet wird: Die Proteinstruktur wird basierend auf der Funktion abgeleitet (gängige Tools wie RFdiffusion), und die generierte Struktur wird dann in ein inverses Proteinfaltungssprachenmodell (wie ProteinMPNN) eingegeben, um schließlich eine Sequenz zu generieren.
* Wenn natürliche Sprache als Medium verwendet wird: Beispielsweise werden mit der im Forschungspapier „Ein textgesteuertes Proteindesign-Framework“ beschriebenen Methode natürliche Sprache und Proteinsequenzen durch vergleichendes Lernen in einem übergeordneten Raum ausgerichtet. Anschließend kann mithilfe natürlicher Sprache direkt in diesem übergeordneten Raum eine Proteinsequenz generiert werden.
Reihenfolge → Struktur
In der Sequenz-zu-Struktur-Richtung ist AlphaFold zweifellos das klassischste Modell. Warum brauchen wir also immer noch Proteinsprachenmodelle für die Strukturvorhersage?Der Hauptgrund ist: schnell.
Der Hauptgrund für die Langsamkeit von AlphaFold liegt darin, dass die MSA-Suche (Multiple Sequence Alignment) große Datenbanken nur mit der CPU durchsucht. GPU-Beschleunigung ist zwar möglich, die tatsächliche Beschleunigung ist jedoch noch geringer. Darüber hinaus erfordert AlphaFold während des Faltvorgangs einen Template-Matching-Prozess, der ebenfalls sehr zeitaufwändig ist. Der Ersatz dieser beiden Module durch ein Protein-Sprachmodell kann den Strukturvorhersageprozess deutlich beschleunigen. Aktuellen Forschungsergebnissen zufolge ist die Genauigkeit der Strukturvorhersage auf Basis von Protein-Sprachmodellen in den meisten Bewertungskriterien jedoch immer noch geringer als die des AlphaFold-Modells.
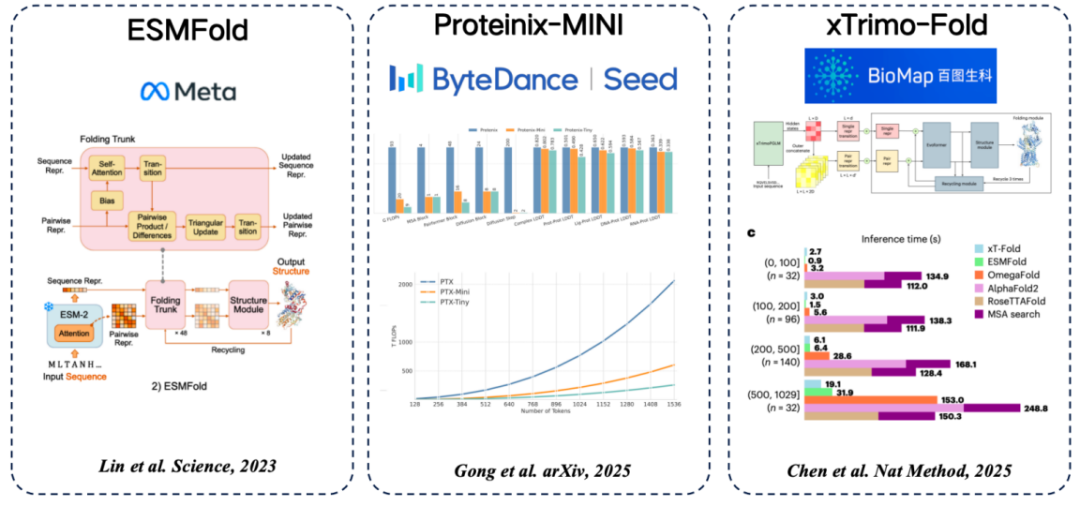
Es gibt mehrere gängige Proteinsprachenmodelle von der Sequenz bis zur Struktur.Die gängige Idee, aus Proteinsprachenmodellen extrahierte Merkmale zu verwenden, um MSA zu ersetzen, wird übernommen:
* ESMFold (Meta): Die erste Methode zur direkten Vorhersage der Proteinstruktur mithilfe eines Proteinsprachenmodells, die eine hohe Genauigkeit erreicht, ohne auf die MSA-Suche angewiesen zu sein.
* Proteinix-MINI (ByteDance): verwendet das Proteinsprachenmodell anstelle von MSA, das ebenfalls sehr schnelle Ergebnisse erzielt und eine Vorhersagegenauigkeit aufweist, die dem AlphaFold 3-Modell nahekommt.
* xTrimo-Fold (Baidu Biosciences): Es nutzt die Funktionen eines Modells mit Hunderten von Milliarden Parametern anstelle von MSA, was die Suche beschleunigt.
Struktur → Reihenfolge
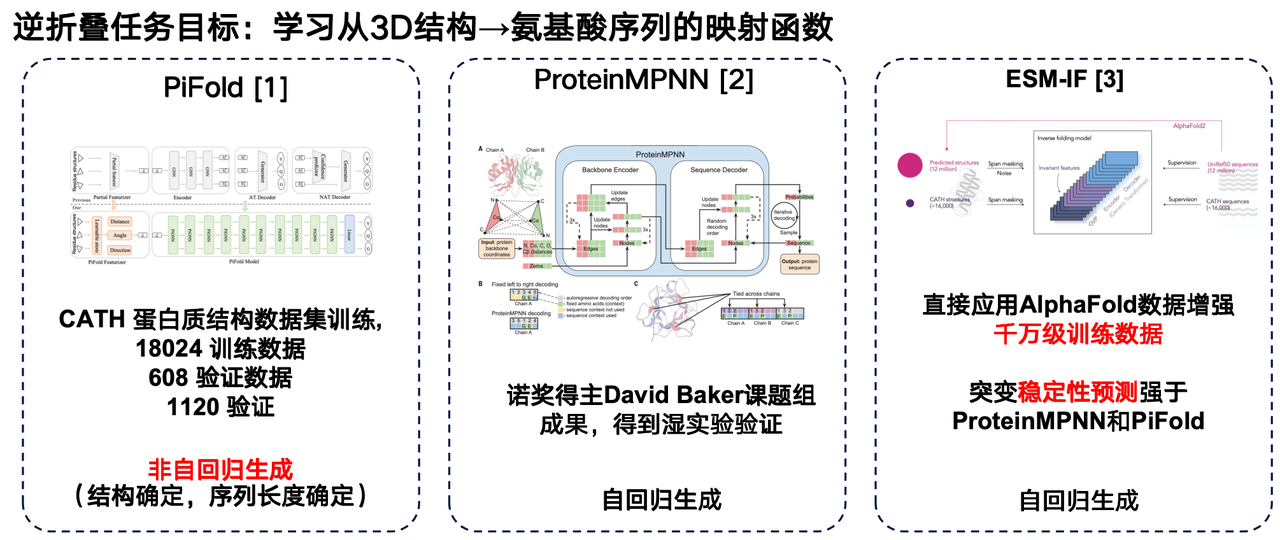
Die Struktur wird auf Grundlage bekannter Funktionen entworfen, aber wie lässt sie sich im Labor synthetisieren?Wir müssen es auch in eine Aminosäuresequenz umwandeln, was das zuvor erwähnte „inverse Faltungssprachenmodell“ ist.
Das inverse Faltungssprachenmodell kann als das „inverse Problem“ von AlphaFold betrachtet werden. Im Gegensatz zu AlphaFold, das die 3D-Struktur aus Aminosäuresequenzen vorhersagt, zielt das inverse Faltungsmodell darauf ab, eine Abbildungsfunktion von der 3D-Struktur eines Proteins auf seine Aminosäuresequenz zu erlernen.
Ich möchte mehrere Arbeiten auf diesem Gebiet vorstellen: Die erste Arbeit ist das PiFold-Modell des Forschungsteams der Westlake University. Eine wesentliche Neuerung seiner Architektur ist die Verwendung einer nicht-autoregressiven Generierungsmethode.
Das zweite Modell ist ProteinMPNN, das von der Forschungsgruppe von David Baker entwickelt wurde. Als eines der am häufigsten verwendeten inversen Faltungsmodelle verwendet es die autoregressive Generierung, um einzelne Proteinstrukturen durch Graph-Neural-Networks zu kodieren und dann nacheinander Aminosäuresequenzen zu generieren.
Auch Metas ESM-IF stellt eine bedeutende Weiterentwicklung dar. Sein Highlight liegt in der Nutzung der umfangreichen Strukturdaten, die von AlphaFold vorhergesagt wurden, um die entsprechenden dreidimensionalen Strukturen für Millionen von Proteinsequenzen einheitlich vorherzusagen und so einen extrem großen Trainingssatz zu erstellen. Die Trainingsdaten von ESM-IF umfassen Millionen von Einheiten, und die Modellanzahl übersteigt 100 Millionen Parameter. Auf dieser Grundlage führt das Modell nicht nur Reverse-Folding-Aufgaben durch, sondern zeigt auch eine starke Leistung bei der Vorhersage der Mutationsstabilität.
Mehrere Ansätze zur Verbesserung von Proteinsprachenmodellen
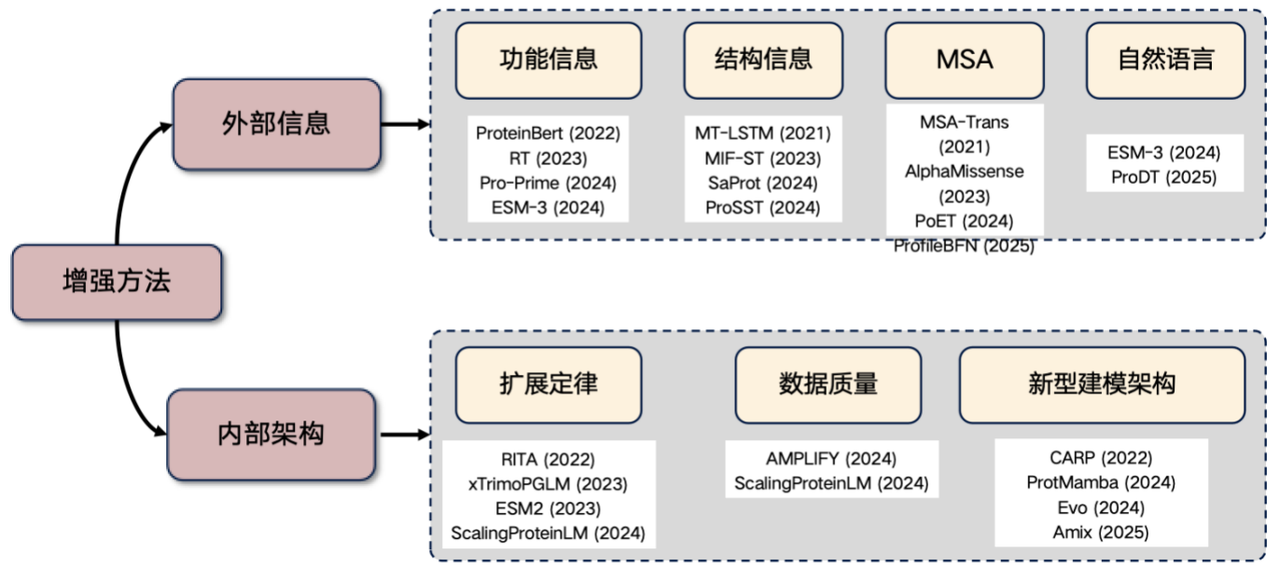
Abschließend möchte ich noch auf eine derzeit sehr beliebte Forschungsrichtung eingehen: die Verbesserung des Protein-Sprachmodells. Wenn Sie in diesem Bereich forschen möchten, können Sie von den folgenden Ideen ausgehen:Externe Informationen einführen und interne Strukturen verbessern.
1. Externe Informationen einbringen
* Funktionale Informationen: Beispielsweise die Eingabe von Merkmalen wie Temperatur und pH-Wert in den Transformer. Diese Informationen können explizit oder durch erlernte Aktionen in die Modelleingabe integriert werden, um die Leistung des Proteinsprachenmodells zu verbessern.
* Strukturinformationen: Einführung dreidimensionaler Struktur- oder strukturierter Sequenzinformationen.
* MSA-Informationen: Multiple Sequence Alignment (MSA) ist eine sehr nützliche Art von Informationen. Ihre Einführung in das Sprachmodell kann die Leistung oft erheblich verbessern.
* Informationen in natürlicher Sprache: In den letzten Jahren haben einige Studien versucht, Informationen in natürlicher Sprache einzubeziehen, aber diese Richtung wird noch erforscht.
2. Verbessern Sie die interne Architektur
* Skalierungsgesetz: Die Leistungsverbesserung wird durch eine deutliche Erhöhung der Anzahl der Modellparameter und der Größe der Trainingsdaten erreicht.
* Verbessern Sie die Datenqualität: Reduzieren Sie Rauschen in den Daten und verbessern Sie die Genauigkeit.
* Erforschung neuer Architekturen: wie CARP-, ProtMamba- und Evo-Architekturen.
In den letzten Jahren hat sich die Nutzung von Proteinstrukturinformationen zur Verbesserung der Modellleistung zu einer wichtigen Forschungsrichtung entwickelt.
Eine der frühesten repräsentativen Studien ist die 2021 erschienene Arbeit „Learning the protein language: Evolution, structure, and function“, die zeigte, wie Strukturinformationen genutzt werden können, um die Fähigkeiten von Proteinsprachmodellen zu verbessern. Anschließend schlug das SaProt-Modell einen cleveren Ansatz vor: Es verknüpft das Aminosäurevokabular des Proteins mit 20 virtuellen Strukturvokabularen, die von Foldseek für Proteinstrukturen generiert wurden, und erzeugt so letztendlich ein kombiniertes Vokabular von 400 (20 × 20) Wörtern. Dieses Vokabular wurde verwendet, um ein maskiertes Sprachmodell zu trainieren, das eine hervorragende Genauigkeit erreichte.
Unser Team trainierte außerdem unabhängig ein multimodales Vortrainingsmodell ProSST für Proteinsequenz und -struktur. Dieses Modell erreicht eine diskrete Darstellung struktureller Informationen, indem es die kontinuierliche Struktur des Proteins in diskrete Token (2.048 verschiedene Token) umwandelt.
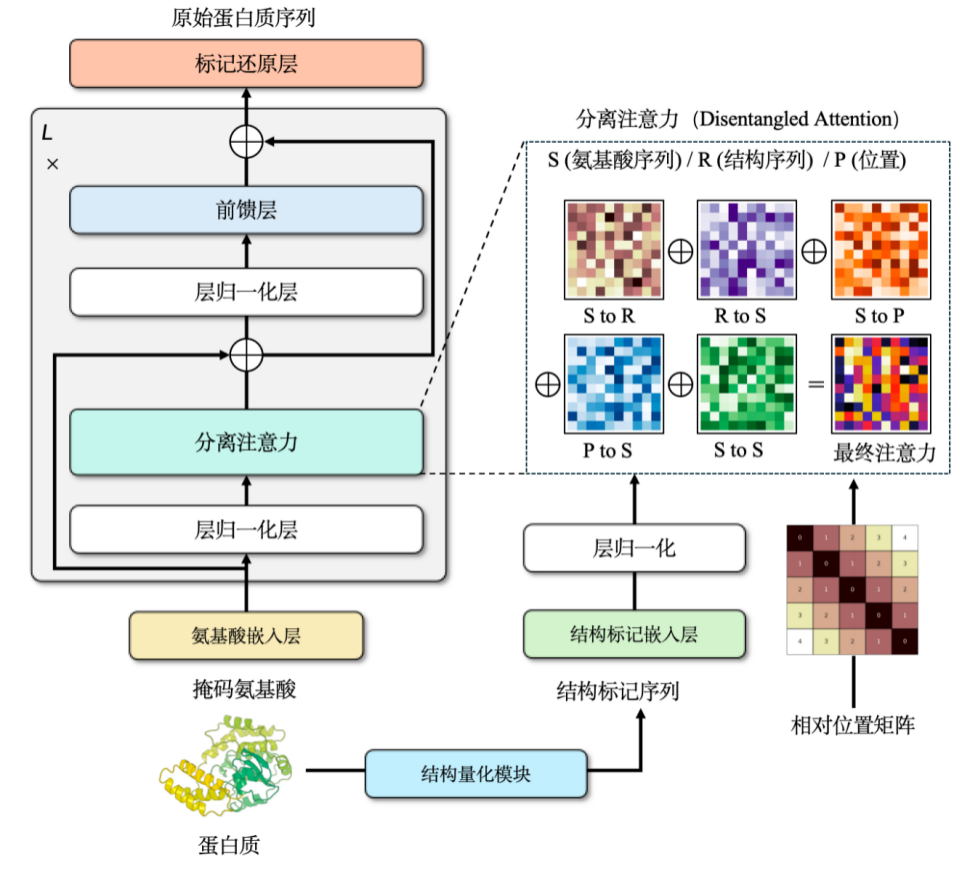
Die Einbeziehung struktureller Informationen in Proteinsprachenmodelle kann die Modellleistung erheblich verbessern. Dabei kann jedoch ein Problem auftreten: Werden die von AlphaFold vorhergesagten Strukturdaten direkt für das Training verwendet, nimmt der Verlust im Trainingsdatensatz allmählich ab, während der Verlust im Validierungs- oder Testdatensatz allmählich zunimmt.Der Schlüssel zur Lösung dieses Problems liegt in der Regularisierung der Strukturinformationen.Einfach ausgedrückt bedeutet dies, komplexe Daten zu vereinfachen, um sie für die Modellverarbeitung besser geeignet zu machen.
Proteinstrukturen werden typischerweise als kontinuierliche Koordinaten im dreidimensionalen Raum dargestellt. Dies muss durch die Umwandlung in diskrete Folgen ganzer Zahlen vereinfacht werden. Zu diesem Zweck verwendeten wir eine Graph-Neural-Network-Architektur und trainierten sie mit einem Denoising-Encoder. So entstand schließlich ein diskretes Strukturvokabular von etwa 2.048 Token.
Mit Struktur- und Sequenzinformationen,Um beides zu kombinieren, haben wir uns für den Cross-Attention-Mechanismus entschieden.Dadurch kann das modifizierte Transformer-Modell sowohl Aminosäure- als auch Struktursequenzen eingeben. Während der Vortrainingsphase haben wir dieses Modell als Aufgabe zur Entwicklung eines Sprachmodells entworfen.Die Trainingsdaten enthalten mehr als 18,8 Millionen hochwertige Proteinstrukturen mit einer Parametergröße von ungefähr 110 Millionen.Das Modell erzielte zu seiner Zeit Spitzenergebnisse und obwohl es inzwischen von neueren Modellen übertroffen wurde, hatte es zum Zeitpunkt seiner Veröffentlichung immer noch die besten Ergebnisse seiner Klasse.
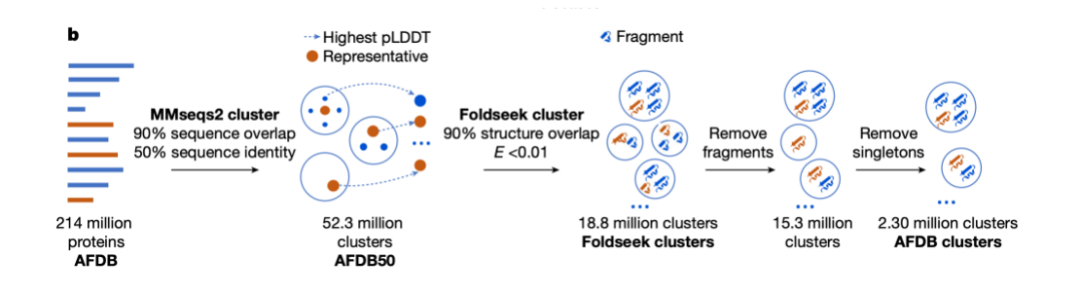
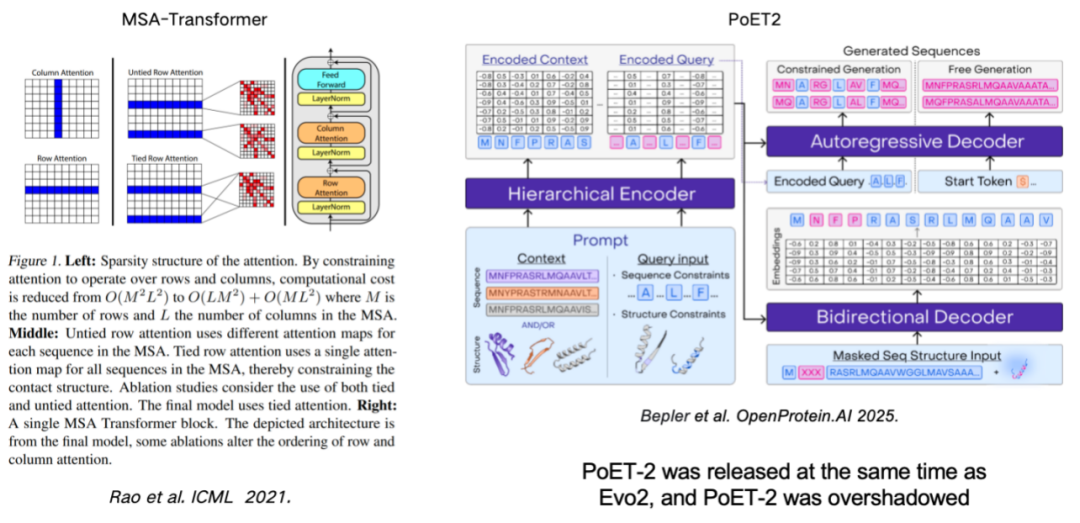
Die Verwendung von MSA (Multiple Sequence Alignment) zur Verbesserung des Proteinsprachenmodells ist ebenfalls ein wichtiges Mittel zur Verbesserung der Modellleistung.Diese Arbeit geht auf den MSA-Transformer zurück, der MSA-Informationen durch die Einführung von Zeilen- und Spaltenregeln effektiv in das Modell integrierte. Das kürzlich veröffentlichte PoET2-Modell verwendet einen hierarchischen Encoder zur Verarbeitung von MSA-Informationen und integriert diese in eine Full-Pass-Modellarchitektur. Nach umfangreichem Training zeigte es eine hervorragende Leistung.
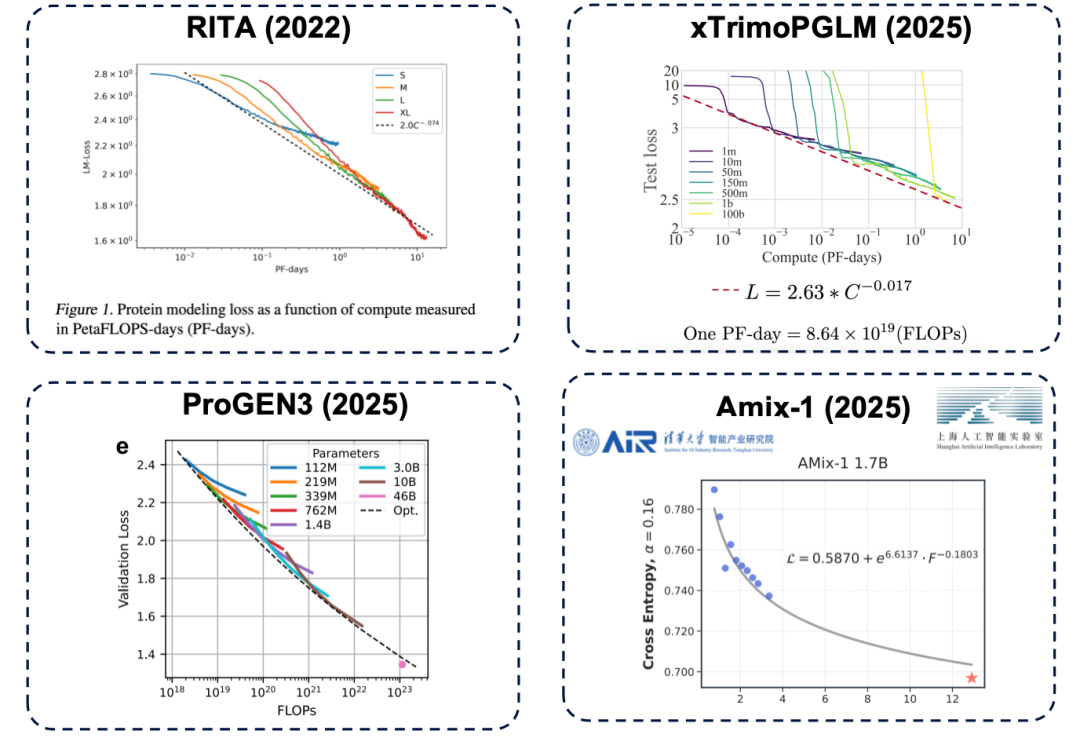
Das Gesetz der Skalierung: Ist ein größeres Modell immer leistungsfähiger?
Das sogenannte Skalierungsgesetz stammt aus der Verarbeitung natürlicher Sprache. Es offenbart ein universelles Gesetz:Die Modellleistung wird sich mit zunehmender Parameterskala, Trainingsdatenmenge und Rechenressourcen weiter verbessern.
Die Parametergröße ist ein entscheidender Faktor für die Bestimmung der Obergrenze der Modellleistung. Reicht die Anzahl der Parameter nicht aus, gerät die Leistung des Modells selbst bei höheren Rechenressourcen (vereinfacht ausgedrückt: höheren Kosten) ins Stocken. Dieses Prinzip gilt auch für Proteinsprachenmodelle und wurde durch zahlreiche Studien bestätigt, darunter repräsentative Arbeiten wie RITA, xTrimoPGLM, ProGEN3 und Amix-1.
* RITA-Modell: entwickelt von der Oxford University, der Harvard Medical School und LightOn AI.
* xTrimoPGLM-Modell: Es wurde vom Baitu Bioscience-Team entwickelt und skaliert die Modellparameter auf ungefähr 100 Milliarden.
* ProGEN3-Modell: entwickelt vom Profluent Biotech-Team.
* Amix-1-Modell: Vorgeschlagen vom Institute of Intelligent Industries der Tsinghua-Universität und dem Shanghai Artificial Intelligence Laboratory, verwendet es eine Bayessche Flussanpassungsnetzwerkarchitektur und verfügt außerdem über ein Expansionsgesetz.
Das zuvor erwähnte „Skalierungsgesetz“ bezieht sich auf den Vortrainingsprozess. In der Proteinforschung geht es jedoch letztlich um die Durchführung nachgelagerter Aufgaben. Dies wirft die Frage auf:Ist eine verbesserte Leistung vor dem Training zwangsläufig hilfreich für nachgelagerte Aufgaben?
Bei der xTrimoPGLM-Auswertung stellte das Forschungsteam fest, dass bei etwa 44%-Downstream-Aufgaben tatsächlich eine positive Korrelation zwischen „besserer Leistung vor dem Training und stärkerer Downstream-Leistung“ besteht.
Gleichzeitig zeigte das Amix-1-Modell emergente Fähigkeiten bei Strukturvorhersageaufgaben. Dabei handelt es sich um Aufgaben, bei denen ein kleines Modell das Problem überhaupt nicht lösen kann, die Leistung sich aber plötzlich deutlich verbessert, wenn die Parametergröße des Modells einen bestimmten kritischen Punkt überschreitet. In diesem Experiment war dieses Phänomen bei Strukturvorhersageaufgaben besonders ausgeprägt, wo die Leistungsverbesserung eine „klippenartige rote Linie“ aufwies, wenn die Parametergröße den kritischen Punkt überschritt.
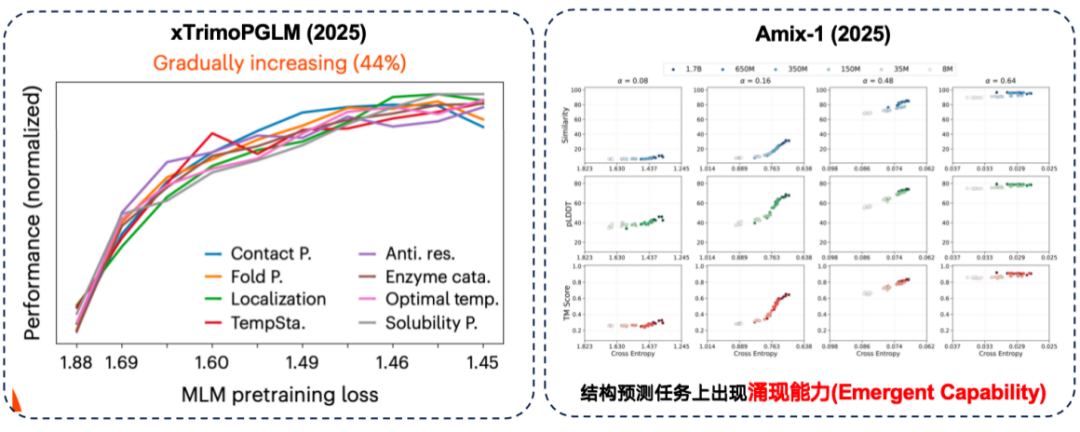
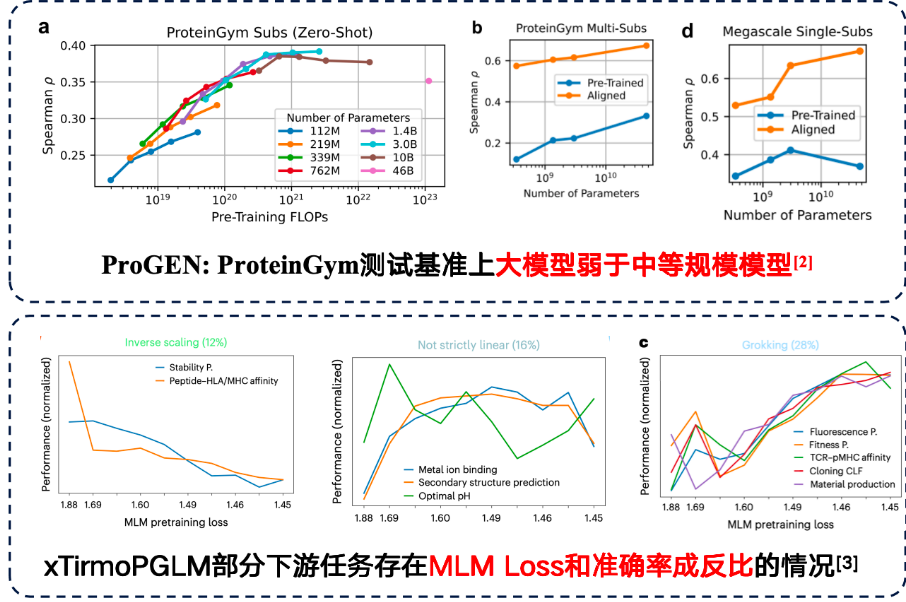
Bei manchen Aufgaben können große Modelle jedoch tatsächlich eine bessere Downstream-Leistung bringen.Bei nachgelagerten Aufgaben wurde jedoch auch ein inverses Skalierungsgesetz entdeckt.Das heißt, je kleiner das Modell, desto besser ist die Leistung.
Studien haben gezeigt, dass eine bloße Erhöhung der Anzahl der Modellparameter die Ergebnisse nicht verbessert, wenn die Trainingsdaten selbst stark verrauscht sind. Daher sollte der Datenqualität mehr Aufmerksamkeit geschenkt werden. Bei der Vorhersage von Proteinmutationen im ProteinGym-Benchmark schnitten mittelgroße Modelle sogar genauer ab. Darüber hinaus entdeckte das Entwicklerteam von xTirmoPGLM auch einige Fälle nicht-positiver Korrelation, bei denen die Leistung vor dem Training nicht mit der Leistung der nachfolgenden Aufgabe übereinstimmte.
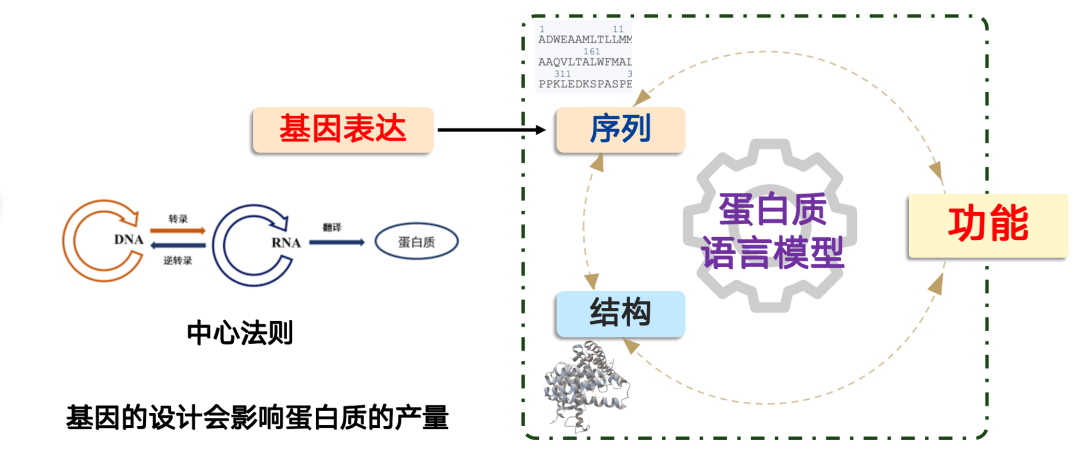
Genomische Modellierung: Vom DNA-Design zur Optimierung der Proteinausbeute
Das Problem, das das Genommodell löst, lautet: Wie produzieren wir ein Protein?
In der synthetischen BiologieDie Herstellung eines Proteins folgt dem zentralen Dogma der Molekularbiologie: „DNA → RNA → Protein“.In Zellen wird dieser Prozess durch den Zellkörper gesteuert, und wir können ihn durch die Gestaltung von Genen abschließen. Der Schlüssel liegt jedoch darin, dass die Gengestaltung die Proteinproduktion direkt beeinflusst.
In der Praxis stoßen wir häufig auf Situationen, in denen ein Protein zwar eine hervorragende funktionelle Leistung aufweist, sein Expressionsniveau jedoch aufgrund eines schlechten genetischen Designs extrem niedrig ist und den Anforderungen einer Industrialisierung oder großtechnischen Anwendung nicht gerecht wird. In diesem Fall können KI-Modelle eine Rolle spielen.
Die Aufgabe des KI-Modells besteht darin, direkt aus Proteinsequenzen abzuleiten, wie DNA-Sequenzen entworfen und deren Produktion gesteigert werden können. Das von unserem Team vorgeschlagene Modell, ProDMM, basiert auf einer Vortrainingsstrategie und besteht aus zwei Phasen:
In der ersten Phase werden durch gemeinsames Vortraining Darstellungen von Proteinen und DNA erlernt. Die Eingaben umfassen Protein- und DNA-Sequenzen, und ein Sprachmodell wird mithilfe einer Transformer-Architektur trainiert. Ziel ist es, gleichzeitig Darstellungen von Protein-, Codon- und DNA-Sequenzen zu erlernen. In der zweiten Phase werden generative Aufgaben anhand nachgelagerter Aufgaben trainiert, beispielsweise vom Protein zur codierenden Sequenz (CDS). Aus einem gegebenen Protein kann eine DNA-Sequenz generiert werden.
* Papieradresse:Li M, Ren Y, Ye P, et al. Nutzung einer einheitlichen multimodalen Sequenzmodellierung zur Aufdeckung der Protein-DNA-Interdependenz[J]. bioRxiv, 2025: 2025.02. 26.640480.
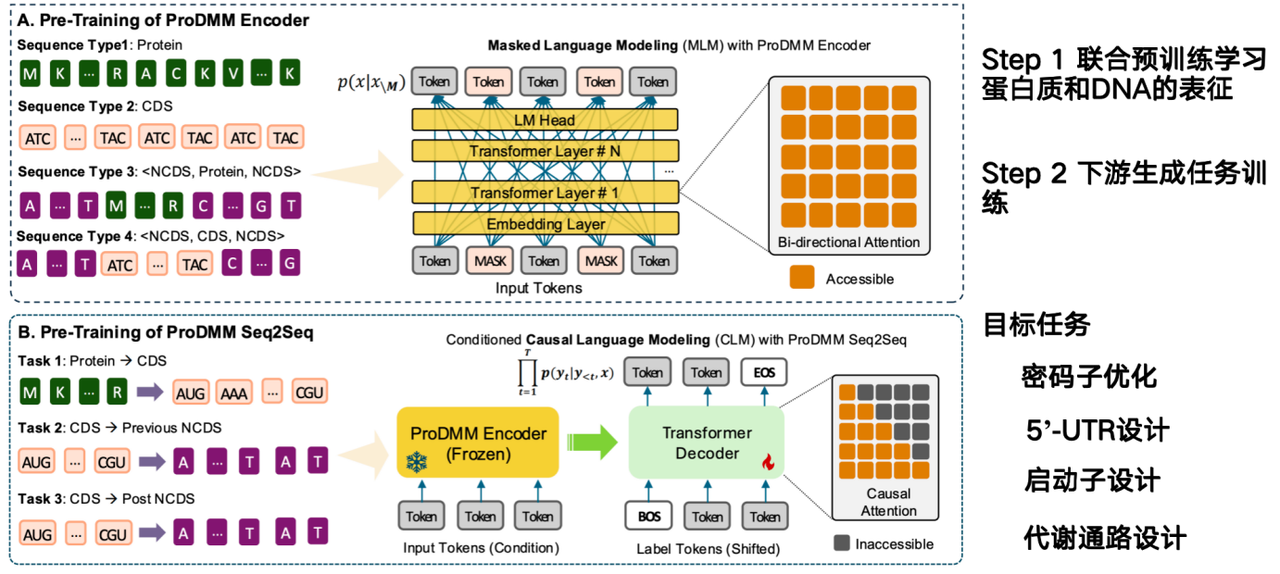
Das Ziel des Projekts „Von Codons zu nicht-kodierender DNA“ (NCDS) besteht darin, die Codon-Optimierung, das 5'-UTR-Design, das Promotor-Design und das Stoffwechselweg-Design abzuschließen.
Die Entwicklung von Stoffwechselwegen umfasst die koordinierte Arbeit mehrerer Proteine innerhalb eines Gens zur Synthese eines bestimmten Produkts. Wir müssen die Produkte des gesamten Stoffwechselwegs optimieren, eine Aufgabe, die sich besonders für genomische Modelle eignet, da Proteinmodelle nur für ein einzelnes Protein optimieren und kontextunabhängig sind. Eine wesentliche Herausforderung für genomische Modelle besteht jedoch darin, die Wechselwirkungen innerhalb der zellulären Umgebung zu berücksichtigen. Dies stellt derzeit ihre größte Herausforderung dar.
Über Dr. Li Mingchen
Gastredner dieser Diskussionsrunde ist Li Mingchen, Postdoktorand in Hong Liangs Forschungsgruppe am Institut für Naturwissenschaften der Shanghai Jiao Tong University. Er promovierte in Informatik und Technologie und erwarb einen Bachelor of Science in Mathematik an der East China University of Science and Technology. Sein Forschungsschwerpunkt liegt auf dem Vortraining von Proteinsprachenmodellen und deren Feinabstimmung.
Er hat den Titel „Shanghai Outstanding Graduate“, das National Scholarship und die Goldmedaille der Shanghai Division des „Internet+“-Wettbewerbs für Innovation und Unternehmertum für College-Studenten gewonnen. Er hat insgesamt 10 SCI-Artikel als Erstautor/Co-Erstautor/korrespondierender Autor in Zeitschriften und Konferenzen wie NeurIPS, Science Advances, Journal of Cheminformatics und Physical Review Research veröffentlicht und war an der Veröffentlichung von 10 SCI-Artikeln beteiligt.
Erhalten Sie mit einem Klick hochwertige Papiere und ausführliche Interpretationsartikel im Bereich AI4S von 2023 bis 2024 ⬇️









