Command Palette
Search for a command to run...
Das Team Von David Baker Schlägt in Science Einen Neuen Ansatz Zur Entwicklung Von Proteinen Zur Bindung Ungeordneter Regionen Vor, Der Speziell Auf Nicht Medikamentös Behandelbare Ziele abzielt.

Da die meisten Krankheiten direkt mit einer gestörten Proteinfunktion zusammenhängen, spielen Proteine eine Schlüsselrolle bei der Arzneimittelentwicklung. Bei der Entwicklung neuer Medikamente nutzen Forscher Proteine häufig als zentrale Angriffspunkte, sodass Medikamente an teilweise stabile Proteine binden und so in den Krankheitsverlauf eingreifen können.Allerdings bleibt es eine Herausforderung, Medikamente gezielt auf intrinsisch ungeordnete Proteine (IDPs) auszurichten, denen es an klar definierten Struktur-, Sequenz- und Konformationspräferenzen mangelt.
Die traditionelle Methode des Antikörper-Targetings basiert hauptsächlich auf der hochspezifischen Bindungsfähigkeit von Antikörpern an spezifische Proteine, um die Erkennung und Regulierung von Zielproteinen zu erreichen. Dieser Targeting-Weg erfordert jedoch nicht nur viele experimentelle Schritte, sondern ungeordnete Antigene werden nach der Injektion auch leicht abgebaut und sind unwirksam. DaherProteine mit intrinsisch ungeordneten Regionen (IDRs), die mehr als 50% im Proteom ausmachen, werden im Allgemeinen als „nicht medikamentös behandelbare“ Ziele angesehen und wurden nie für die Arzneimittelentwicklung verwendet.
In diesem Zusammenhang schlugen David Baker, ein herausragender Computerbiologe, der 2024 den Nobelpreis für Chemie erhält und Direktor des Institute for Protein Design an der University of Washington ist, und sein Team eine Proteindesignstrategie namens Logos vor.Basierend auf der Induced Fit-Bindungsstrategie wurden Bindungsproteine entwickelt, die sich an 39 ungeordnete Zielaminosäuresequenzen anpassen können.Diese Studie generierte ein spezialisiertes erweitertes Proteinrückgrat und verallgemeinerte es anschließend mithilfe des RFdiffusionsmodells. Das Rückgrat enthält speziell für Peptid-Repeat-Sequenzen entwickelte Taschen, die es der entwickelten Binder-Zielpeptid-Vorlage ermöglichen, ungeordnete Proteinbereiche universell zu erkennen. Dies bedeutet, dass mehr Proteine als Angriffspunkte für die Entwicklung neuer Medikamente dienen und so die Krebs- und Alzheimerforschung beschleunigen könnten.
Die entsprechenden Forschungsergebnisse wurden in Science unter dem Titel „Design of intrinsically disordered region binding proteins“ veröffentlicht.
Forschungshighlights:
*Erstellen Sie eine für die allgemeine Erkennung geeignete Vorlagenstrukturbibliothek, um eine Bindungsanpassungskonformationsinduktion für jede Zielsequenz zu erreichen.
* Entwicklung von Bindungsproteinen für 18 synthetische Peptidsequenzen und 21 natürlich ungeordnete Regionen (IDRs) mit großer Diversität und therapeutischem Potenzial, die in der Lage sind, ungeordnete Regionen krebsassoziierter extrazellulärer Rezeptoren anzusprechen und die Proteinlokalisierung innerhalb der Zellen voranzutreiben.

Papieradresse:
https://www.science.org/doi/10.1126/science.adr8063
Folgen Sie dem offiziellen Konto und antworten Sie mit „Natural disordered protein“, um das vollständige PDF zu erhalten
Weitere Artikel zu den Grenzen der KI:
Generierung einer Template-Bibliothek: Universelle Peptididentifizierung
Diese Studie kombiniert physikalische Designmethoden und Deep-Learning-Designmethoden als Lösung für das IDR-Bindungsproblem. Eingeschränkt durch die Inkompatibilität von Peptideinheiten mit heterogenen Zielsequenzen,Die Studie begann mit verschiedenen Wiederholungsproteinstrukturen und verwendete das Diffusionsmodell, um die Aminosäurebindungstaschen in verschiedenen Wiederholungseinheiten neu zu organisieren und sie in verschiedene Aminosäuren und Konformationsvorlagen zu differenzieren.Dies ermöglicht eine breitere Erkennung von Sequenzen.
Um Peptide in natürlich ungeordneten Proteinen zu identifizieren, wurde im Rahmen der Studie zunächst eine Backbone-Vorlagenbibliothek erstellt. Diese Vorlagenbibliothek weist zwei Merkmale auf:
*Jede Matrizenstruktur sollte in der Lage sein, die gestreckte Peptidkettenkonformation zu „umhüllen“ und eine große Anzahl von Möglichkeiten für Wechselwirkungen wie Wasserstoffbrücken und dichte Packung zu bieten, wodurch eine hochspezifische Erkennung der Zielsequenz erreicht wird.
* Die Template-Struktur ist breit gefächert und kann mit jeder Zielsequenz übereinstimmen, so dass mindestens ein Template eine definierte und geeignete Bindungskonformation induzieren kann.
Der Prozess der Generierung einer Backbone-Vorlagenbibliothek ist in drei Schritte unterteilt: Backbone-Generierung, Protein-aktive Taschenspezialisierung und Protein-aktive Taschenassemblierung.
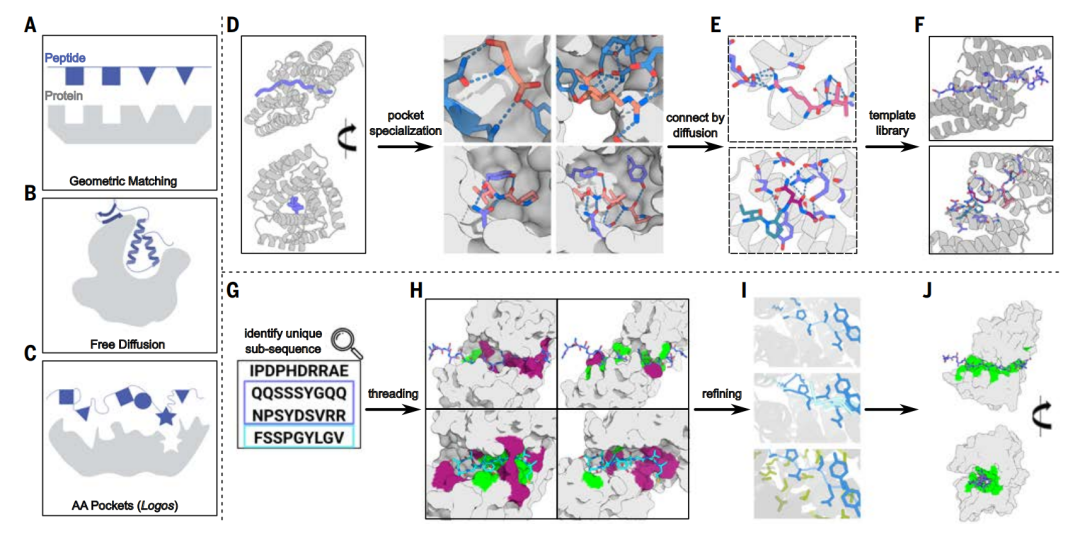
Gerüstgenerierung
Während der Phase der Rückgratgenerierung konzentrierte sich das Forschungsteam auf mehrere erweiterte Konformationen, anstatt sich auf die Polyprolin-II-Konformation zu beschränken, da die Polyprolin-II-Konformation hauptsächlich in prolinreichen Peptiden vorkommt.
In der gestreckten Konformation weisen die Seitenketten der Aminosäuren abwechselnd in entgegengesetzte Richtungen, was mit den Eigenschaften einer Wiederholung aus zwei Aminosäuren übereinstimmt.Die Forscher verwendeten die Rosetta-Designmethode, um eine Reihe von Dipeptid-Wiederholungssequenzen zu entwerfen.Sie umfassen LK, RT, YD, PV und GA (alles einbuchstabige Abkürzungen für Aminosäuren) und sind so konzipiert, dass sie sich mit diesen Peptidsegmenten in unterschiedlichen gestreckten Konformationen verwickeln und an sie binden, sodass jede sich wiederholende Einheit mit einer Dipeptideinheit interagiert.
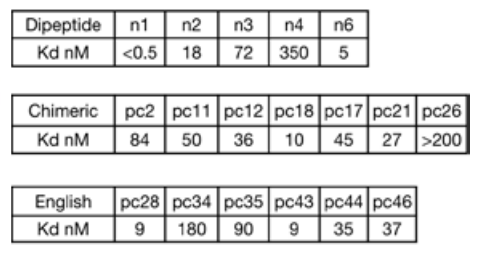
Anschließend charakterisierten die Forscher diese mit vier Wiederholungseinheiten ausgestatteten Versionen der Bindungsproteine durch Fluoreszenzpolarisationsexperimente. Die Ergebnisse zeigten, dass sie eine nanomolare Bindungsfähigkeit für die Wiederholungspeptide LK und PV aufwiesen; für die polareren Peptide RT und YD war die Bindungsfähigkeit jedoch schwächer, und für das hochflexible GA wurde überhaupt kein Bindungssignal nachgewiesen.
Taschenspezialisierung
Beim Festlegen der aktiven Proteintasche verwendeten die Forscher Diffusionsmodellierung, um die Tasche fein abzustimmen und eine genauere Übereinstimmung mit der spezifischen Zielpeptidsequenz zu erreichen.
Um die Effizienz der Template-Anpassung zu verbessern, verfeinerten die Forscher die entworfene Bindungstasche, indem sie die Anzahl der interagierenden Wiederholungseinheiten von vier auf fünf erhöhten und gleichzeitig die Übereinstimmung mit der Zielsequenz verbesserten. Dieser Ansatz erhöhte auch die Affinität zwischen den Zielstrukturen. Die vier bis neun Aminosäuren, die jede gegabelte Wasserstoffbrücke der Seitenkette zwischen dem Wiederholungsprotein und dem Peptidrückgrat umgeben, blieben unverändert, während die hydrophoben Wechselwirkungen zwischen den entworfenen Bindungsproteinen diversifiziert wurden.
Der Vorteil dieser Strategie besteht darin, dass die geometrischen Konfigurationsanforderungen für Wasserstoffbrücken strenger sind. Im Vergleich dazu bietet die unpolare hydrophobe Stapelung eine höhere räumliche Freiheit. Daher ist es im Design effizienter, Wasserstoffbrücken direkt in einer Schablonenform zu erhalten, als Wasserstoffbrücken immer wieder von Grund auf neu zu erstellen.
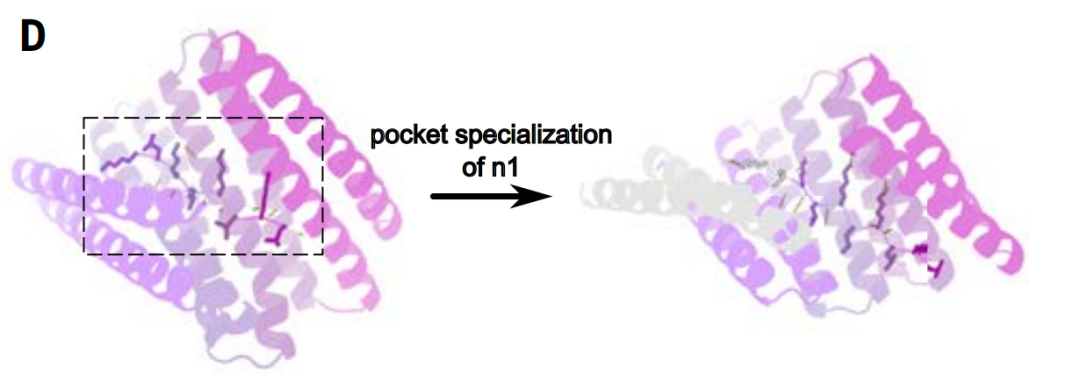
Die neu erweiterte fünfte Wiederholungsstruktur ist hellgrau dargestellt
Taschenmontage
Während des TaschenmontageschrittsDie Forscher verwendeten das RFdiffusionsmodell, um Schnittstellen zwischen Taschen zu erstellen, was zu einer insgesamt starren Struktur führte und eine Vorlage für die Montage der Bindungstaschen zu einem neuen Rückgrat erzeugte.Die verschiedenen Taschen in der Vorlage sind in unterschiedlicher Reihenfolge und Geometrie angeordnet, um mit dem Peptidziel in einer Reihe erweiterter Konformationen zu interagieren, was eine allgemeinere Erkennung nicht-repetitiver Sequenzen ermöglicht.
Nach der Generierung chimärer Proteinmodelle, die mit chimären Peptidzielen interagieren, lokalisierte die Studie die Bindungstaschen parametrisch und verband sie mittels Radiofrequenzdiffusion. Mit diesem Ansatz generierte die Studie 70 Designvorschläge für sieben chimäre Ziele. Die Charakterisierung durch Split-Luciferase-Häufigkeits- und Biolayer-Interferometrie-Experimente ergab, dass bei durchschnittlich nur zehn getesteten Designs pro Ziel sechs der sieben Ziele eine Bindung im zweistelligen nanomolaren Bereich erreichten.
Um die Größe der Vorlagenbibliothek zu erweitern und einen größeren Bereich von Sequenzen abzudecken, verwendete die Studie die Taschenassemblierungstechnologie, um 36 chimäre Rückgrate zu konstruieren, die Taschen enthalten, die polare Reste erkennen, und erzeugte 1.000 Vorlagen, die aus einem entworfenen Bindungsprotein und einem entsprechenden Peptidrückgrat bestehen, in dem die Aminosäuren in der Peptidkonformation mit den entworfenen Taschen im Bindungsprotein übereinstimmen können.
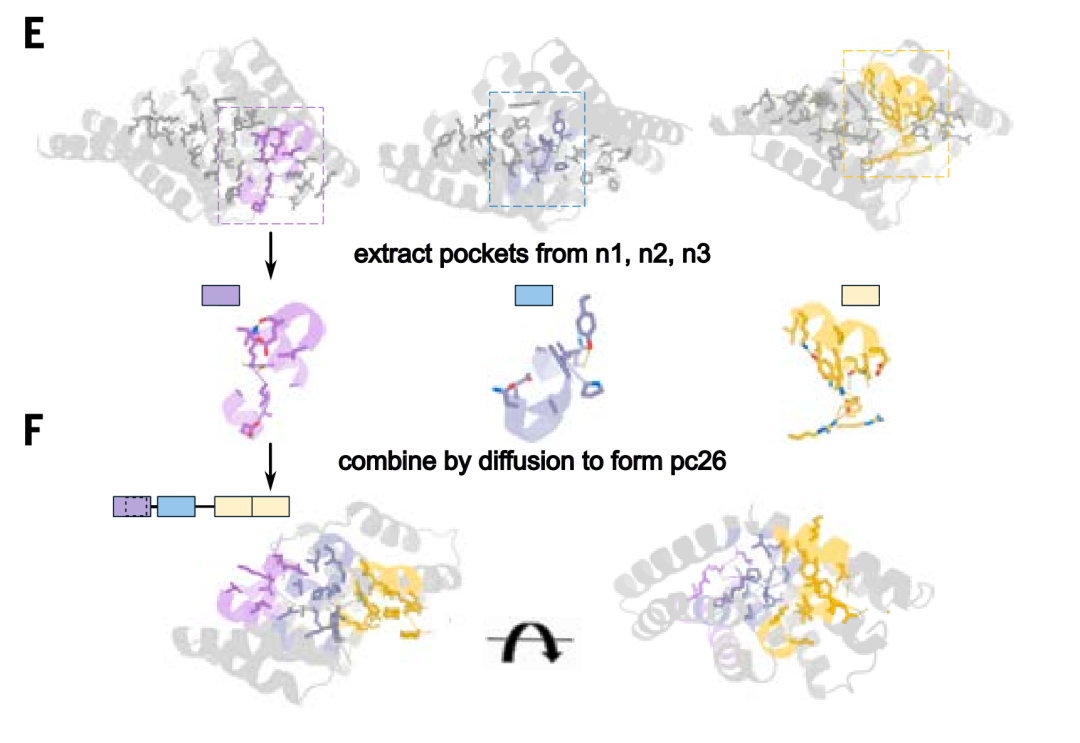
Design und Optimierung von IDR-Bindungsproteinen
Nach dem Aufbau einer Vorlagenbibliothek fügten die Forscher natürlich ungeordnete Regionen in diese ein und nutzten sie zur Erzeugung von Bindeproteinen, die an nicht-repetitive synthetische Sequenzen und jedes natürlich unstrukturierte Ziel binden können. Dieser Schritt gliedert sich in zwei Teile: Threading-Matching und Strukturverfeinerung.
Thread-Matching: Bestimmen des kompatibelsten Sequenzfragment-Vorlagen-Paares
Beim Threading-Matching wird die Zielsequenz in das Rückgrat jeder Vorlage eingefädelt, um das kompatibelste Sequenzfragment zu identifizieren, das mit der Vorlage gepaart werden kann.
Generell gibt es bei IDP oder IDR eine große Anzahl möglicher Peptide, die als Zielmoleküle verwendet werden können. Um das Peptid mit dem größten Zielpotenzial in IDR zu finden,Die Studie eliminierte zunächst Peptide mit geringer Sequenzkomplexität und Peptide mit mehreren engen Übereinstimmungen im Proteom, um Kreuzreaktionen mit Bindern solcher Ziele zu verhindern.Nach dem lokalen Backbone-Resampling durch Zuordnung der einzigartigen Sequenzfragmente der verbleibenden Aminosäuren zum Ziel-Backbone der Template-Bibliothek,In der Studie wurde das auf Deep Learning basierende Protein-Sequenz-Design-Tool ProteinMPNN verwendet, um die Sequenz des Bindungsproteins zu optimieren und diese anhand der Übereinstimmung zwischen dem entworfenen Bindungsprotein und der Zielsequenz sowie der Konsistenz zwischen dem AF2-Vorhersagewert und dem Modell zu bewerten.
In Fällen, in denen die AF2-Metriken nicht optimal waren, wurde RFdiffusion verwendet, um das Backbone für ein bestimmtes Ziel anzupassen. Anschließend wurde Threaded Matching verwendet, um Binder für therapeutisch relevante IDPs, IDRs und IDP-Fragmente zu generieren, wodurch durchschnittlich 28 Designs pro Ziel generiert wurden.
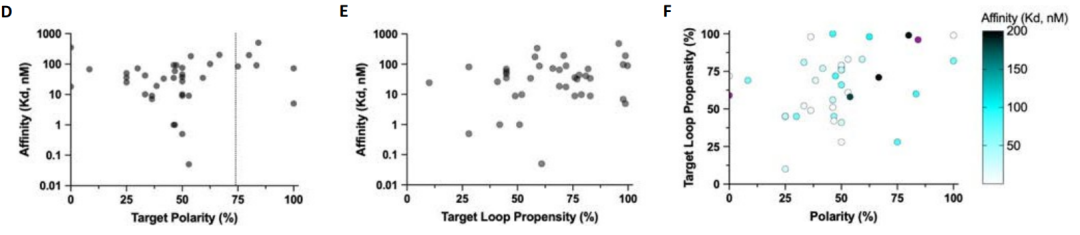
Strukturoptimierung: Verbesserung der Übereinstimmung zwischen Bindeprotein und Zielpeptid
Die beste Übereinstimmung wurde außerdem optimiert, um die Übereinstimmung zwischen dem entworfenen Bindungsprotein und dem Zielpeptid zu verbessern.Für die Studie wurde die Dissoziationskonstante des DYNA_1b1-Bindungsproteins und Dynorphins zum Testen ausgewählt und die Radiofrequenzdiffusion für die höchste Trefferquote bei synthetischen Zielen optimiert.Die Ergebnisse zeigten, dass 45 der 48 Designs im Screening-Test eine starke Affinität zeigten und nur die Dissoziationskonstanten von 6 Designs eine schwache Affinität zeigten.
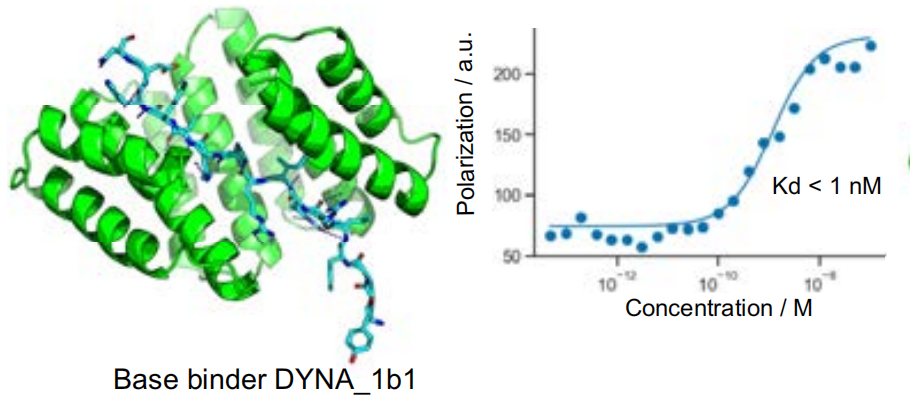
Validierung der Orthogonalität zwischen Dynorphinstruktur und Bindungsprotein
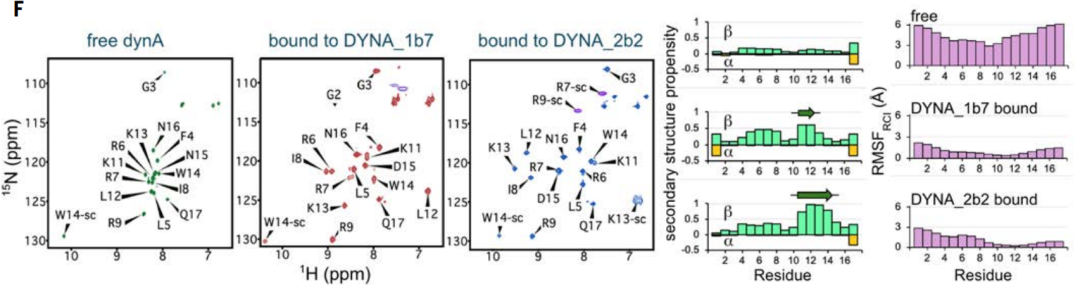
Um die Veränderungen in der Dynorphinstruktur während der Bindung zu überprüfen, wurden die Kernspinresonanzspektren (NMR) von isotopenmarkiertem Dynorphin A in Lösung erfasst, wenn es ungebunden, an DYNA_1b1 gebunden und mit höherer Affinität an DYNA_2b2 gebunden war.
Aus den NMR-Ergebnissen ging hervor, dass freies Dynorphin A intrinsisch ungeordnet ist, die im entworfenen Rückgrat enthaltenen Regionen jedoch bei der Bindung geordnet werden.Für beide gebundenen Komplexe zeigten die NMR-Daten erweiterte Bindungszustandskonformationen, die mit dem entworfenen Modell übereinstimmen und die Wirksamkeit der Dynorphinwirkung bei der Induktion ungeordneter Proteine und Peptide in nicht-native Konformationen bestätigen.
Um das Optimierungspotenzial von Logos zu erkunden, wählten die Forscher einen Dynorphin-Binder, DYNA_1b1, mit einer Bindungskonstante (Kd) von etwa 1 nM. Die Forscher optimierten die am besten bewerteten Designs mittels RF-Diffusion. Von den 48 Designs zeigten 45 mittels BLI-Screening eine starke Bindung bei einer Konzentration von 5 nM, wobei sechs Designs Kd-Werte von ≤100 pM aufwiesen, gemessen mittels BLI. Fluoreszenzpolarisationsmessungen von zwei der optimierten Designs (DYNA_2b1 und DYNA_2b2) ergaben Kd-Werte unter 60 pM bzw. 100 pM, wie in Abbildung B unten dargestellt.
Hinweis: Dynorphin ist ein Peptidligand des Kappa-Opioid-Rezeptors (KOR), der mit chronischen Schmerzen in Verbindung gebracht wird.
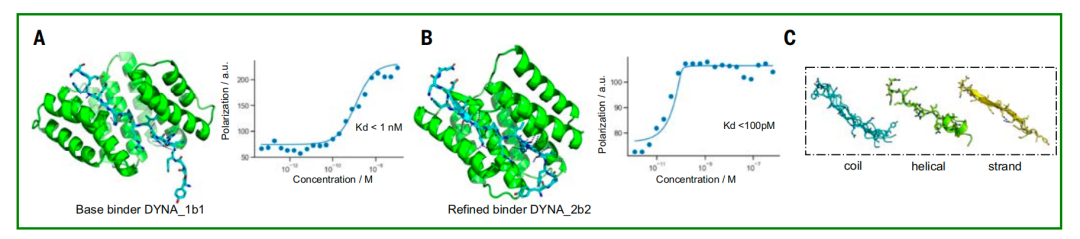
In den ursprünglichen und optimierten Designs von Dynorphin A zeigte das Peptid mehrere Konformationen, darunter Zufallsknäuel, partielle β-Strang-Strukturen und partielle α-Helix-Strukturen, wie in Abbildung C oben dargestellt. Obwohl Dynorphin A und B eine Sequenzähnlichkeit von 62% aufweisen, kreuzen sich ihre jeweiligen Bindungsproteine nicht und binden nur an ihre jeweiligen Zielmoleküle. Gleichzeitig ist die Co-Kristallstruktur des an Dynorphin A gebundenen Proteins DYNA_1b7 in hohem Maße konsistent mit dem computergestützten Designmodell, insbesondere an der Kernbindungsschnittstelle (Abbildungen DE oben). NMR-Daten bestätigten außerdem, dassDas ursprünglich ungeordnete Skelett des Dynorphins A wird nach der Bindung an das entworfene Protein geordnet, was erneut die Wirksamkeit des induzierten Anpassungsmechanismus bestätigt (Abbildung F oben).
Überprüfen Sie die Funktionalität und Orthogonalität von Bindungsproteinen
Die Studie verwendete Immunpräzipitationsmodelle des WASH-Komplexes und des PER-Komplexes. Der WASH-Komplex umfasst WASH, FAM21, CCDC53, SWIP und WASHC2. Tests zeigten, dass FAM21_1b1 den gesamten WASH-Komplex aus Zelllysaten extrahierte.
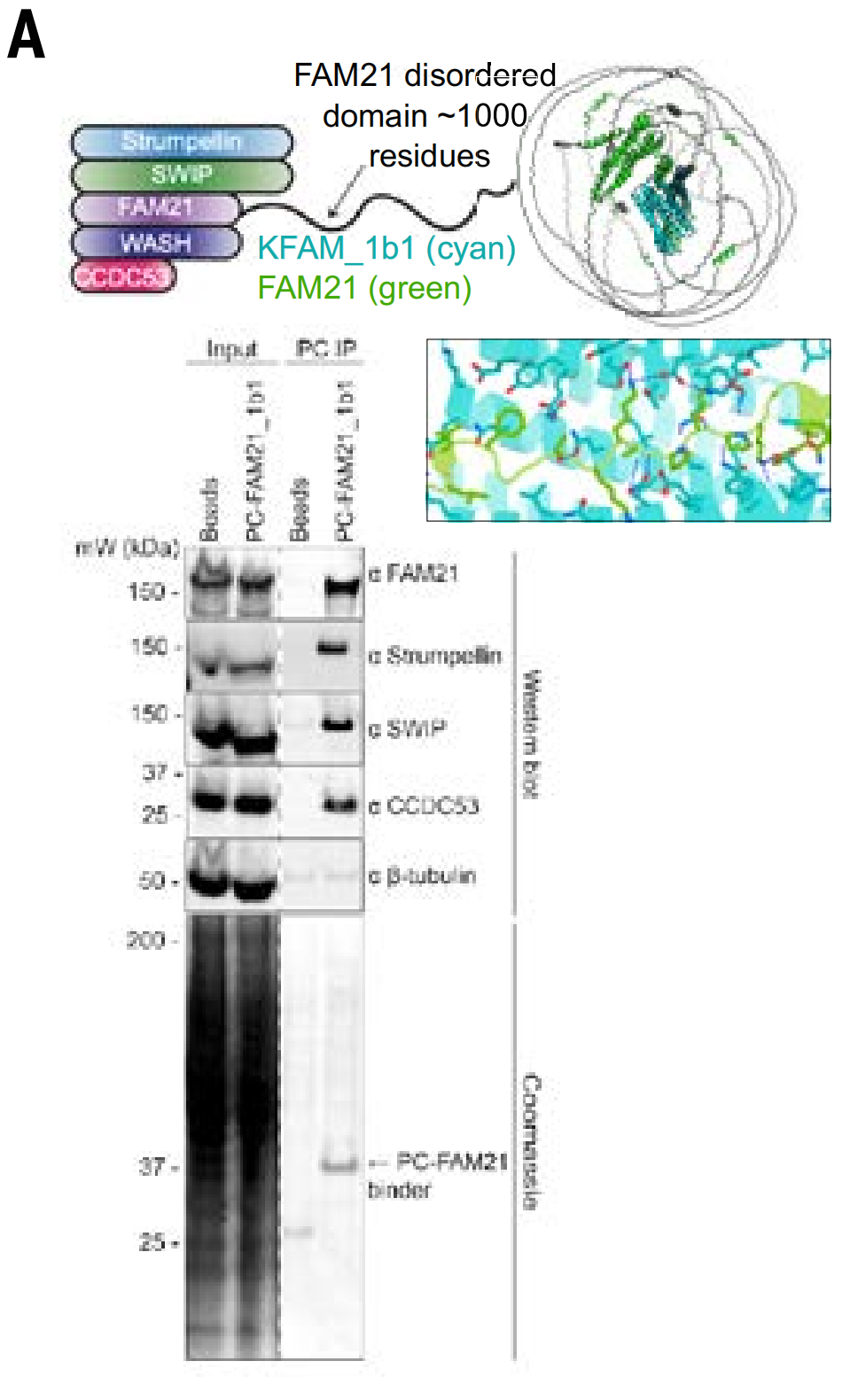
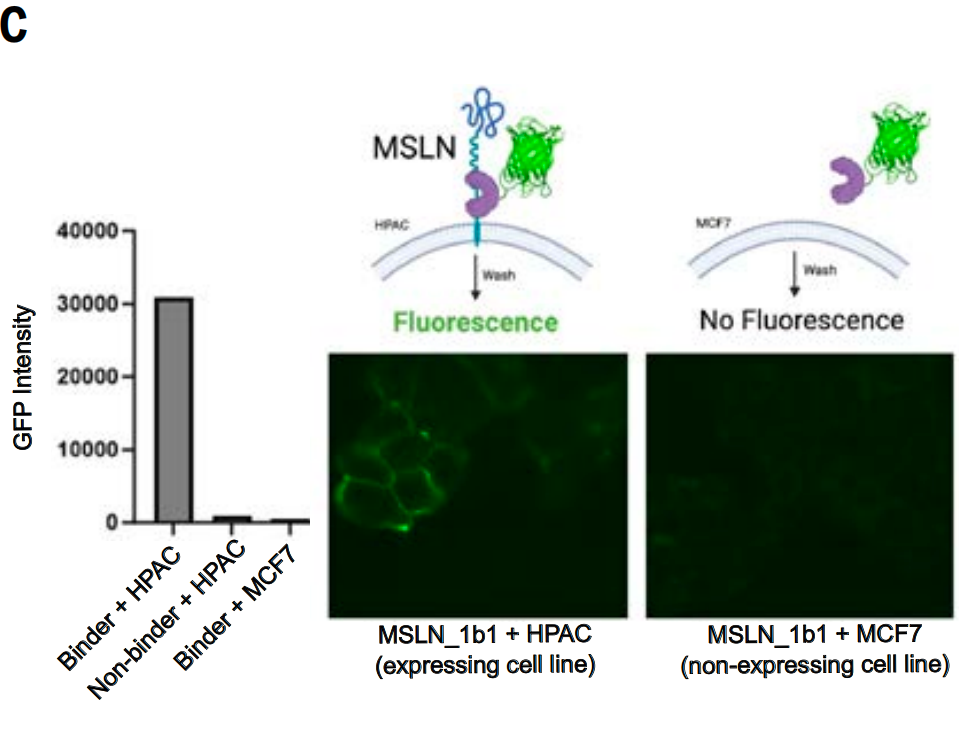
Darüber hinaus untersuchten sie auch, ob ein für die MSLN-Juxtamembranregion entwickeltes Bindungsprotein (MSLN_1b1) spezifisch an Zellen binden kann, die dieses Ziel exprimieren (da die Proteasespaltung in dieser Region die distalere extrazelluläre Domänenregion als Ziel weniger geeignet macht).
Hinweis: Mesothelin (MSLN) ist ein Zelloberflächen-Glykoprotein, das bei vielen Krebsarten hochreguliert ist und daher in der Tumor-zielgerichteten Therapie viel Aufmerksamkeit auf sich gezogen hat.
Die Forscher fusionierten grünes fluoreszierendes Protein (GFP) mit MSLN_1b1 und inkubierten es mit Zellen, die MSLN exprimierten (menschliche Pankreasadenokarzinom-Zelllinie HPAC) und einer Zelllinie, die MSLN nicht exprimierte (Brustkrebszelllinie MCF7 der Michigan Cancer Foundation). Als Kontrolle nahmen sie außerdem ein GFP-Fusionsprotein auf, das nicht an MSLN bindet.
Fluoreszenzmikroskopie zeigte, dass das GFP-MSLN_1b1-Fusionsprotein an Zellverbindungen in HPAC-Zellen aggregierte, was mit den Lokalisationsmerkmalen von MSLN übereinstimmt. Dieses Phänomen wurde in MCF7-Kontrollzellen nicht beobachtet. Darüber hinaus zeigte das Kontrollbindungsprotein kein Bindungssignal in HPAC-Zellen, wie in Abbildung C unten dargestellt. MSLN_1b1 erkennt und bindet daher spezifisch an MSLN auf der Zelloberfläche.
KI-gesteuert, eröffnet neue Perspektiven für das Protein-Targeting
KI wird derzeit zunehmend in der Proteinforschung eingesetzt und treibt die Forschung in eine neue Phase der „multitechnischen Parallelität“ voran. Neben David Bakers Team haben auch die Teams von George M. Burslem und Ophir Shalem an der University of Pennsylvania bahnbrechende Durchbrüche in der Proteinforschung erzielt. Dieses Team entwickelte eine „Protein-Editing“-Technologie, bei der ein Split-Intein-System erfolgreich eingesetzt wird, um die Aminosäuresequenz von Proteinen nach der Synthese in lebenden Säugetierzellen direkt zu modifizieren. Dies ist das erste Mal, dass nicht-standardmäßige Aminosäuren und chemische Markierungen (Biotin, Fluorophore) präzise in endogene Proteine eingebaut wurden. Die Forschungsergebnisse wurden in Science unter dem Titel „Intracellular protein editing enables incorporation of noncanonical residues in endogenous proteins“ veröffentlicht.
Papieradresse:
https://www.science.org/doi/10.1126/science.adr5499
Darüber hinaus entwickelte ein chinesisch-internationales Team unter der Leitung von Gao Caixia vom Institut für Genetik und Entwicklungsbiologie der Chinesischen Akademie der Wissenschaften und Li Guotian von der Huazhong Agricultural University, aufbauend auf den Proteindesign-Arbeiten von David Bakers Team, AiCE, eine universelle Protein-Engineering-Methode auf Basis inverser Faltungsmodelle. Mithilfe dieser KI-gesteuerten Proteindesign-Strategie optimierten sie erfolgreich acht Proteinklassen, darunter Deaminasen und Nukleasen, und entwickelten einen neuartigen Baseneditor. Die Forschungsarbeit mit dem Titel „Advancing protein evolution with inverse folding models integrating structural and evolutionary constraints“ wurde in Cell veröffentlicht.
Papieradresse:
https://www.cell.com/cell/abstract/S0092-8674(25)00680-4
Von der Live-Cell-Editierung bis hin zu neuroprotektiven Therapien, von Glykosylierungsinnovationen bis hin zu KI-gestütztem Multi-Chain-Design – mit der kontinuierlichen Weiterentwicklung der KI in der Biomedizin nutzen Teams weltweit beispiellos vielfältige Ansätze, um die biomedizinischen Herausforderungen im Zusammenhang mit natürlich ungeordneten Proteinen zu bewältigen. Die Erforschung natürlich ungeordneter Regionen durch das Forschungsteam wird neue Therapieansätze für Krankheiten wie Krebs und Alzheimer schaffen.
Referenzlinks:








