Command Palette
Search for a command to run...
Datenentrauschung/biologische Signalverstärkung/Dropout-Linderung, Deep-Learning-Modell SUICA Ermöglicht Vorhersage Der Genexpression an Jeder Position in Räumlichen Transkriptomschnitten

Die Gruppe um Professor Zheng Yinqiang von der Universität Tokio und die Gruppe um Professor Ding Jun von der McGill University haben gemeinsam eine Methode zur Modellierung räumlicher Transkriptomdaten namens SUICA vorgeschlagen. SUICA ist ein Deep-Learning-Modell, das auf impliziten neuronalen Repräsentationen (INR) und Graph-Autoencodern basiert. SUICA reduziert mithilfe von Graph-Autoencodern die Dimensionalität hochdimensionaler räumlicher Transkriptomdaten und modelliert anschließend mithilfe impliziter neuronaler Repräsentationen die Koordinaten der räumlichen Transkriptomdaten und die entsprechenden Genexpressionen. Dadurch lässt sich die Genexpression an jeder Position im räumlichen Transkriptomschnitt vorhersagen.Die Ergebnisse zeigen, dass räumliche Transkriptomdaten, die von SUICA verarbeitet werden, eine höhere Qualität, weniger Rauschen und stärkere biologische Signale aufweisen können.
Die entsprechenden Ergebnisse wurden für ICML 2025 unter dem Titel „SUICA: Learning Super-high Dimensional Sparse Implicit Neural Representations for Spatial Transcriptomics“ ausgewählt.

Papieradresse:
https://go.hyper.ai/C6Zcl
konzentrieren Sie sich auf 「HyperAI "Offizielles WeChat-Konto. Antworten Sie im Backstage-Bereich mit „SUICA“, um das vollständige PDF zu erhalten.
Weitere Artikel zu den Grenzen der KI:
https://go.hyper.ai/owxf6
Was sind räumliche Transkriptomdaten?
Bei den Daten der räumlichen Transkriptomik (ST) handelt es sich um eine hochdimensionale Informationsmatrix, die gleichzeitig „Genexpressionsniveaus“ und „räumliche Koordinaten“ im selben Gewebeschnitt aufzeichnet.Im Vergleich zur herkömmlichen Panorama-Gewebebildgebung (WSI), die nur morphologische Strukturen darstellen kann, oder zur konventionellen Transkriptomik, die die Genexpression nur quantifizieren kann, aber die Richtung verliert, verknüpft die räumliche Transkriptomik „welche Gene exprimiert werden“ mit „wo im Gewebe sie sich befinden“ und zeichnet eine funktionelle Karte der Interaktion zwischen Zellstatus und Mikroumgebung im Gewebe und wird so zu einer neuen Datenform, die Histologie und molekulare Omics verbindet.
Warum besteht die Notwendigkeit, räumliche transkriptomische Daten zu verbessern?
Obwohl die räumliche Transkriptomik beispiellose räumlich aufgelöste molekulare Erkenntnisse gebracht hat, sind die Daten aus der realen Welt immer noch durch drei große Engpässe begrenzt:
① Auflösungs-Kosten-Widerspruch:Je dichter die Sonden und je höher die Sequenzierungstiefe, desto schneller steigen die experimentellen Kosten (z. B. sind die Sequenzierungsexperimentkosten von Stereo-Seq höher als $4.000/cm²) und der Probendurchsatz.
2 Signalschwachheit und Rauschen:Die Anzahl der an jedem Detektionspunkt erfassten mRNAs ist begrenzt, und eine Null-Expansion ist schwerwiegend, wodurch es leicht passieren kann, dass Gene mit geringer Häufigkeit oder wichtige regulatorische Gene übersehen werden.
3 Plattformübergreifende Heterogenität:Verschiedene Plattformen weisen erhebliche Unterschiede in der physikalischen Anordnung der Sonden, der Sequenzierungstiefe und dem Hintergrundrauschen auf, was die Integration mehrerer Proben oder mehrerer Experimente direkt behindert.
Zu den rechnergestützten Verbesserungsmethoden zählen die Rekonstruktion mit Superauflösung, die Tiefenentrauschung und das Auffüllen fehlender Werte. Mit diesen Methoden lässt sich Folgendes erreichen, ohne dass sich der experimentelle Aufwand erhöht (oder nur geringfügig erhöht):
(a) Vorhersage der Genexpression an Stellen, die nicht sequenziert wurden;
(b) Wiederherstellung der wahren Genexpression, die aufgrund technischer Einschränkungen nicht nachgewiesen werden kann, und Verbesserung der Empfindlichkeit beim Nachweis unterschiedlich exprimierter Gene und räumlich variabler Gene;
(c) Erstellen Sie standardisierte Merkmalsdarstellungen, die plattformübergreifend vergleichbar und gemeinsam nutzbar sind.
Dadurch wird eine genauere, umfangreichere und skalierbarere Datengrundlage für die Analyse der Zellkommunikation, die Annotation von Krankheitszonen, die Entdeckung von Arzneimittelzielen, die gemeinsame Multi-Omics-Modellierung und die KI-gestützte pathologiegestützte Diagnose geschaffen und das Potenzial der räumlichen Transkriptomik-Technologie in der Grundlagenforschung und der klinischen Transformation in großem Umfang freigesetzt.
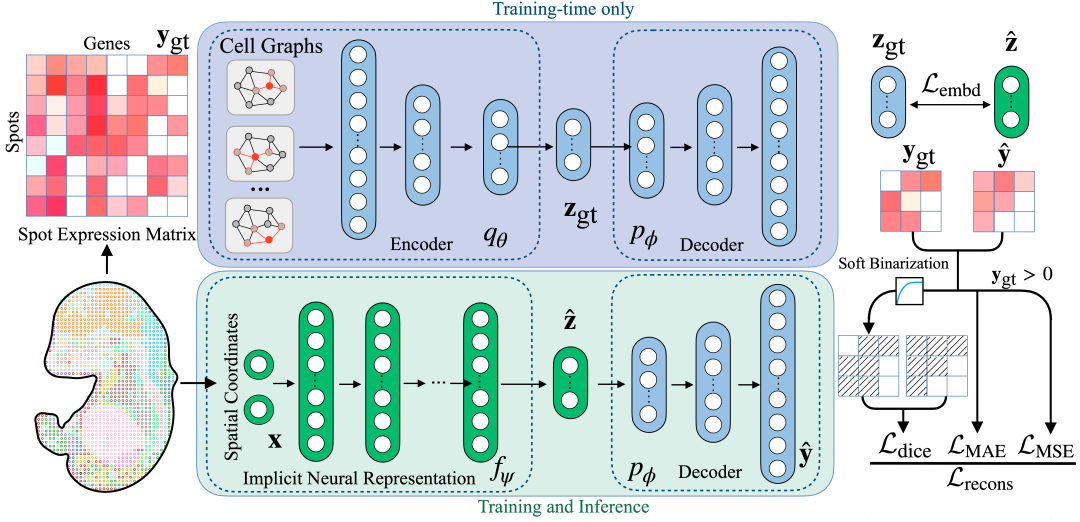
SUICA: Ein einheitliches Modell basierend auf impliziter neuronaler Repräsentation und Graph-Autoencoder
Herausforderungen bei der Modellierung räumlicher transkriptomischer Daten mithilfe impliziter neuronaler Repräsentationen
Die Modellierung räumlicher transkriptomischer Daten ist mit zahlreichen Herausforderungen verbunden:
Zunächst werden die Originaldaten rasterartig in der räumlichen Dimension verteilt.In Bezug auf die Gendimension beträgt die Zahl Tausende bis Zehntausende und bildet eine „ultrahochdimensionale, extrem spärliche und verrauschte“ Matrix. Die hohe Dropout-Rate schwächt die wichtigsten biologischen Signale und verschlimmert den Mangel an statistischer Aussagekraft.
Zweitens besteht bei vorhandenen räumlichen Transkriptomik-Plattformen ein grundlegender Kompromiss zwischen Auflösung und Kosten.——Je dichter die Sonden und je tiefer die Sequenzierung, desto exponentieller steigen die Kosten, was es schwierig macht, gleichzeitig eine Auflösung auf Zellebene und große Probengrößen zu erreichen.
Drittens müssen beim Versuch, diskrete räumliche Transkriptompunkte mithilfe impliziter neuronaler Darstellung in kontinuierliche Expressionsfelder zu interpolieren, zwei große technische Schwierigkeiten gleichzeitig gelöst werden: Erstens übersteigt die Dimension des Genexpressionraums die herkömmlicher visueller Signale bei weitem, und durch einfaches Erweitern oder Vertiefen des Netzwerks lässt sich der Dimensionsfluch nur schwer beseitigen. Zweitens führt die Nullerweiterung zu einer sehr ungleichmäßigen Verteilung der Eingangssignale, und mit herkömmlichen INRs lassen sich komplexe und nichtlineare räumliche Expressionsmuster nur schwer erfassen.
Abbildungs-Autoencoder: Reduzierung der Dimensionalität hochdimensionaler Raumtranskriptomdaten
Im Vergleich zu herkömmlichen Autoencodern betrachten wir die Datenpunkte in jedem räumlichen Transkriptom zunächst als Graphknoten und erstellen eine Adjazenzmatrix basierend auf der räumlichen Nähe. Anschließend nutzen wir die Graphenfaltung im Encoder, um die ursprüngliche hochdimensionale Genexpression zu falten, den lokalen räumlichen Kontext in die Darstellung einzubeziehen und sie in eine niedrigdimensionale Darstellung zu komprimieren. Auf diese Weise lernen wir die niedrigdimensionale Darstellung hochdimensionaler räumlicher Transkriptomdaten, und die zusätzliche Graphenfaltung kann das spärliche und verrauschte Signal der räumlichen Transkriptomdaten verbessern.
Implizite neuronale Repräsentation: Erstellen einer Zuordnung zwischen Sequenzierungspunktkoordinaten und Genexpression
Nachdem die niedrigdimensionale Darstellung erhalten wurde,Das implizite neuronale Darstellungsnetzwerk erhält die Koordinaten der Erkennungspunkte als Eingabe und lernt die Zuordnung zwischen dem „Punkt“ und seiner entsprechenden niedrigdimensionalen Darstellung.Und die erlernte, vom Modell vorhergesagte niedrigdimensionale Darstellung wird an den Decoder-Teil des Graph-Autoencoders gesendet, wodurch die Funktion der Zuordnung von Koordinaten zur hochdimensionalen Genexpression erreicht wird.
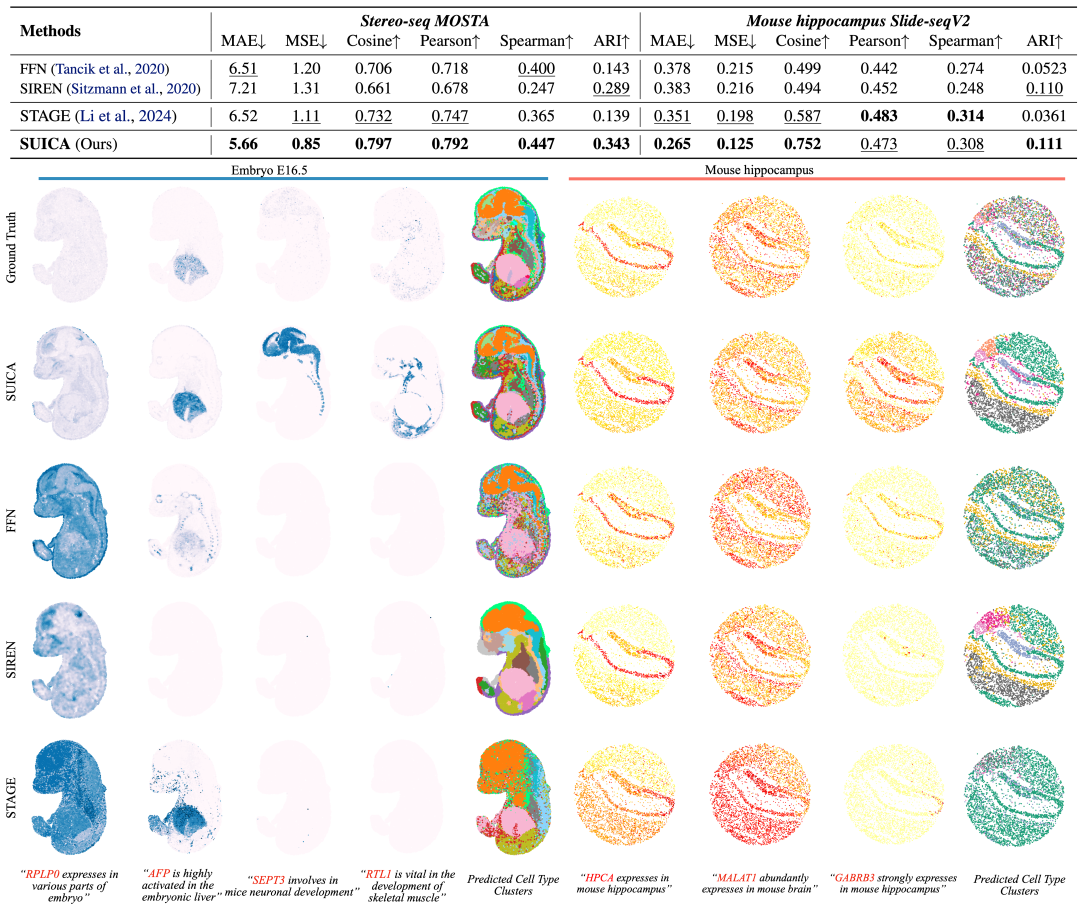
Experimentelle Überprüfung: SUICA kann genauere und biologisch relevantere Vorhersageergebnisse liefern
Wir verwendeten Stereosequenzdaten von Mausembryos und Slidesequenzdaten von Maushirnschnitten für den Benchmark-Vergleich. Bei der Vorhersage unbekannter Punkte (Superauflösung) übertraf SUICA bestehende Modelle und traditionelle implizite neuronale Repräsentationsmodelle, darunter FFN und SIREN, in mehreren Schlüsselindikatoren deutlich. Wir visualisierten den Vorhersageeffekt jeder Methode, und die Ergebnisse zeigten, dass die Vorhersage von SUICA nicht nur das Expressionsmuster von Genen präzise wiederherstellen, sondern auch das Expressionssignal von Genen verstärken kann. Beispielsweise konnte SEPT3, ein Gen, das eine wichtige Rolle bei der Entwicklung des Nervensystems von Mausembryonen spielt, dieses Signal erfolgreich erfassen, obwohl es in der Groundtruth nicht eindeutig war.
Durch Clustering und Kennzeichnung der mit verschiedenen Methoden generierten Ergebnisse stellten wir intuitiv fest, dass die von SUICA generierten Zelltypen den realen Zelltypen am nächsten kommen. Darüber hinaus weisen die von SUICA generierten Zelltypen detailliertere Organ- und Gewebestrukturen im Raum auf.Diese Ergebnisse zeigen, dass SUICA in der Lage ist, biologische Signale zu verstärken und subtile Unterschiede im Zellzustand zwischen verschiedenen Organen und Geweben zu erkennen.
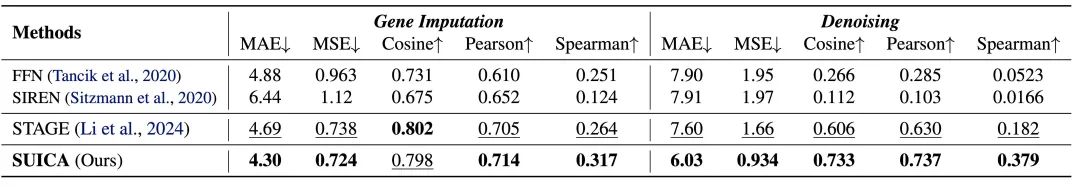
Experimentelle Überprüfung: SUICA kann das Rauschen räumlicher Transkriptomdaten reduzieren und das Dropout-Phänomen lindern
Um die Rauschunterdrückungsfähigkeit (Genimputation) von SUICA und seine Fähigkeit zur Wiederherstellung der wahren Genexpression aus Dropouts (die aus 0 Reads aufgrund von Einschränkungen der Sequenzierungstechnologie resultieren) zu überprüfen, haben wir den räumlichen Transkriptomdaten künstlich Gaußsches Rauschen hinzugefügt oder die Genexpression zufällig auf 0 gesetzt. Im Genimputationsexperiment haben wir 70 % der Genexpression in den Daten zufällig auf 0 gesetzt. Im Experiment zur Rauschunterdrückung der Genexpression haben wir alle negativen Werte auf Null gesetzt, um sicherzustellen, dass die Genexpressionsverteilung nach dem Hinzufügen des Rauschens noch immer der ursprünglichen Genexpressionsverteilung ähnelt.Experimentelle Ergebnisse zeigen, dass SUICA bestehenden Methoden in mehreren Indikatoren überlegen ist, was seine Fähigkeit beweist, Rauschen in räumlichen Transkriptomdaten zu reduzieren und das Dropout-Phänomen zu lindern.