Command Palette
Search for a command to run...
NVIDIA Erzielt Durchbruch Im Proteindesign Auf Atomarer Ebene Und Generiert Proteine Mit Bis Zu 800 Aminosäureresten Mit Hoher Genauigkeit
Es ist bekannt, dass die Entwicklung neuer Proteine mit spezifischen Strukturen und Funktionen großes Anwendungspotenzial in vielen Bereichen wie der Arzneimittelentwicklung und der Biotechnologie bietet. Dieses Ziel zu erreichen, ist jedoch nicht einfach, insbesondere wenn es darum geht, die Beziehung zwischen Proteinsequenz und -struktur zu erfassen, was bei der Entwicklung von Proteinen von Grund auf eine große Herausforderung darstellt.
In der Vergangenheit wurden bei den meisten Methoden die Proteinsequenz und -struktur getrennt entworfen.Beispielsweise muss zuerst die Sequenz generiert und dann gefaltet werden, oder zuerst das Rückgrat entworfen und dann die Sequenz bestimmt werden. Es ist jedoch nach wie vor eine große Herausforderung, die gemeinsame Verteilung von Proteinsequenz und Atomstruktur präzise zu modellieren, um eine präzise Kontrolle der funktionellen Stellen zu erreichen und wichtige Aufgaben des Proteindesigns wie den Entwurf des atomaren Motivgerüsts zu erfüllen. Dies erfordert nicht nur den Umgang mit diskreten Sequenzen und kontinuierlichen Koordinaten, sondern auch die Berücksichtigung des Problems der sich mit der Sequenz ändernden Seitenkettenabmessungen.
In diesem ZusammenhangDas Forschungsteam von NVIDIA und Mila, das Quebec Institute for Artificial Intelligence in Kanada, haben La-Proteina vorgeschlagen.Hierbei handelt es sich um eine Proteindesignmethode auf atomarer Ebene, die auf partiellem Potentialfluss-Matching basiert. Sie kann die explizite Backbone-Modellierung und die Potentialdarstellung jedes Restes mit fester Größe effektiv kombinieren, um Sequenz- und atomare Seitenketteninformationen zu erfassen. Dadurch wird die zentrale Herausforderung der Dimensionsvariabilität der expliziten Seitenkettendarstellung bei der Proteingenerierung gelöst und neue Durchbrüche im Bereich des Proteindesigns erzielt.
Die entsprechenden Forschungsergebnisse wurden auf arXiv unter dem Titel „La-Proteina: Atomistic Protein Generation via Partially Latent Flow Matching“ veröffentlicht.
Forschungshighlights:
* Es wird ein teilweise implizites Flussabgleichs-Framework La-Proteina vorgeschlagen, das für die gemeinsame Generierung von Proteinsequenzen und vollständigen Strukturen auf atomarer Ebene konzipiert ist. Es kombiniert effektiv explizite Hauptkettenmodellierung und eine implizite Darstellung jedes Restes mit fester Größe, um sowohl Sequenz- als auch Seitenketten auf atomarer Ebene zu erfassen.
* In umfangreichen Benchmark-Experimenten erreicht La-Proteina eine SOTA-Leistung bei der bedingungslosen Proteingenerierung und ist in der Lage, vielfältige, gemeinsam gestaltbare und strukturell gültige Proteine im vollständigen atomaren Maßstab mit bis zu 800 Resten zu erzeugen.
* In der Studie wurde La-Proteina erfolgreich auf das indizierte und nicht indizierte Motivgerüstdesign auf atomarer Ebene angewendet, zwei wichtige Aufgaben des bedingten Proteindesigns, und beide zeigten, dass das Modell früheren All-Atom-Generatoren überlegen ist.

Papieradresse:
Weitere Artikel zu den Grenzen der KI:
https://go.hyper.ai/owxf6
Datensatz: Wird zum Trainieren unbedingter Modelle sowie von Merkmalen und Funktionen von Proteindaten verwendet
In dieser Studie wurden zwei Datensätze zum Trainieren unbedingter Modelle verwendet:
Einer davon ist der von Foldseek geclusterte AFDB-Datensatz, der aus der Überprüfung und Clusterung der AlphaFold-Datenbank (AFDB) abgeleitet ist.Die Clusterung kombiniert Sequenz- und Strukturinformationen mit einem anfänglichen Satz von etwa 3 Millionen Einzelproben, die anhand mehrerer Kriterien optimiert wurden: ein durchschnittlicher pLDDT-Score von mindestens 80, eine Proteinlänge von 32 bis 512 Aminosäuren, ein Coiling-Verhältnis von weniger als 50% und maximal 20 aufeinanderfolgende Coiling-Reste. Das Vorhandensein von β-Faltungen war insbesondere erforderlich, um den niedrigen β-Faltungsgehalt in den modellgenerierten Proteinen zu korrigieren.Schließlich wurden etwa 550.000 Proteinproben gewonnen.Dieser Datensatz wurde sorgfältig geprüft, um die Strukturmerkmale der vom Modell generierten Proteine ausgewogener zu gestalten und insbesondere den β-Faltungsgehalt zu verbessern.
Die zweite ist eine angepasste AFDB-Teilmenge für das Training langer Sequenzen.Die Forscher untersuchten Proben von AFDB mit einem durchschnittlichen pLDDT von mindestens 70 und einer Länge zwischen 384 und 896.Nach der Clusterbildung wurden mehr als 4 Millionen Cluster für das Training erhalten.Durch die Konzentration auf längere Proteinproben wird es den Anforderungen des Trainings langer Sequenzen gerecht.
Darüber hinaus enthalten die Proteindaten selbst Sequenzinformationen (20 Resttypen) und 3D-Strukturinformationen, die mithilfe der Atom37-Darstellung einheitlich gespeichert werden. Die Atom37-Darstellung definiert eine standardisierte Obermenge von 37 potenziellen Atomen für jeden Rest. Eine Proteinstruktur mit L Resten kann als Tensor der Form [L, 37, 3] gespeichert werden, und die relevanten Koordinatenuntermengen werden entsprechend dem Typ jedes Rests ausgewählt.
Das Besondere an dieser Standardisierungsmethode ist die einheitliche Speicherung und Darstellung der Strukturinformationen verschiedener Reste. Dies bildet die Grundlage für die einheitliche Verarbeitung der Strukturinformationen verschiedener Reste durch das Modell. Die umfangreichen Dateneigenschaften von AFDB liefern dem Modell umfangreiche Stichproben, die ihm helfen, ein breiteres Spektrum an Proteinsequenzen und Strukturmerkmalen zu erlernen und so die Leistung und Generalisierungsfähigkeit zu verbessern. Durch Training und Experimente mit diesen Daten können verwandte Modelle die Beziehung zwischen Proteinsequenz und -struktur besser erfassen und ein präziseres Design erreichen.
La-Proteina: Innovative Architektur und Trainingsmechanismus eines Proteindesignmodells auf atomarer Ebene
La-Proteina ist ein innovatives Modell für Proteindesign auf atomarer Ebene. Sein Kerndesign basiert auf der „partiellen impliziten Repräsentation“ und zielt darauf ab, die komplexen Herausforderungen bei der Generierung vollständig atomarer Strukturen zu lösen.
Auf der Designebene, angesichts der Herausforderung, All-Atom-Strukturen zu erzeugen und dabei das großflächige Rückgrat, die Aminosäuretypen und die Seitenketten zu berücksichtigen (die Seitenkettenabmessungen variieren je nach Aminosäure),La-Proteina schlägt vor, die Details auf atomarer Ebene und den Resttyp jedes Rests in einem kontinuierlichen latenten Raum fester Länge zu kodieren und gleichzeitig die explizite Rückgratmodellierung durch α-Kohlenstoff-Koordinaten beizubehalten.
Dieses Design bietet mehrere Vorteile: Es vermeidet nicht nur die Schwierigkeiten der gemischten kontinuierlichen Klassifizierungsmodellierung in der Hauptgenerierungskomponente des Modells, wodurch die Methode des vollständigen kontinuierlichen Flussabgleichs die effiziente Generierung verborgener Variablen ermöglicht, sondern kann auch auf der Weiterentwicklung der leistungsstarken Hauptkettenmodellierung basieren. Gleichzeitig ermöglicht die explizite Hauptkettenmodellierung die Festlegung unterschiedlicher Generierungspläne für das globale α-Kohlenstoff-Rückgrat und die Details der Reste auf atomarer Ebene. Dies ist der Schlüssel zu hoher Leistung und verbessert zudem die Skalierbarkeit, sodass das Modell auf große Proteine mit bis zu 800 Resten erweitert werden kann. Dieser hybride Ansatz ist der Hauptgrund für seine Überlegenheit gegenüber dem vollständig impliziten Modellierungsrahmen.
Aus der Kompositionsstruktur, wie in der Abbildung unten dargestellt,Der Kern von La-Proteina besteht aus drei neuronalen Netzwerken: Encoder, Decoder und Denoiser.Alle drei teilen die Kernarchitektur von Transformer, die auf dem Mechanismus der voreingenommenen Aufmerksamkeit basiert.
Der Encoder ist für die Abbildung des Eingabeproteins (mit Sequenz- und Strukturinformationen) auf latente Variablen zuständig. Seine anfängliche Sequenzdarstellung umfasst die ursprünglichen Atomkoordinaten, Seitenketten- und Rückgrat-Torsionswinkel sowie Resttypen, und die anfängliche Paardarstellung umfasst die relative Sequenztrennung, den paarweisen Abstand und die relative Richtung zwischen den Resten. Der Decoder rekonstruiert das vollständige Protein aus den latenten Variablen und den α-Kohlenstoffatomkoordinaten und verarbeitet dabei 8-dimensionale latente Variablen und α-Kohlenstoffatomkoordinaten für jeden Rest. Das Denoiser-Netzwerk wird zur Vorhersage des Geschwindigkeitsfelds verwendet, das Proben von der standardmäßigen Gauß-Referenzverteilung in die Zieldatenverteilung überträgt, und bestimmt die Interpolationszeit direkt in seinem Transformer-Block.
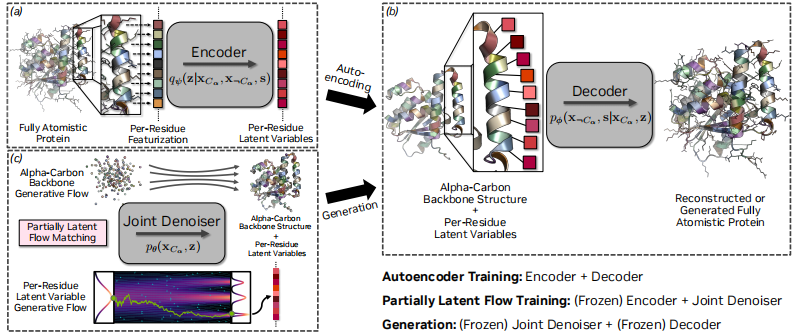
Was die Trainingsmethoden betrifft,La-Proteina verwendet eine zweiphasige Trainingsstrategie.
Die erste Stufe trainiert einen bedingten Variational Autoencoder (VAE): Der Encoder bildet das Eingangsprotein auf latente Variablen ab, und der Decoder rekonstruiert das Protein basierend auf den latenten Variablen und den α-Kohlenstoff-Atomkoordinaten. Der gesamte VAE wird durch Maximierung der β-gewichteten Evidenzuntergrenze (ELBO) optimiert. Für die oben genannten Modellierungsoptionen lässt sich der Rekonstruktionsterm auf den Kreuzentropieverlust der Sequenz und den quadrierten L2-Verlust der Struktur vereinfachen.
Die zweite Stufe optimiert das Flow-Matching-Modell, um die Zielverteilung anzunähern. Das Denoiser-Netzwerk wird durch Minimierung des CFM-Ziels (Conditional Flow Matching) trainiert. Die Verwendung zweier separater Interpolationszeiten tx und tz ist das zentrale Design dieser Stufe. Diese Einstellung ermöglicht unterschiedliche Integrationspläne für die α-Kohlenstoff-Atomkoordinaten und latenten Variablen während der Inferenz und verbessert so die Modellleistung effektiv.
Durch ein solches Design und Training ist La-Proteina in der Lage, die gemeinsame Verteilung von Proteinsequenzen und Allatomstrukturen effizient zu erlernen und bietet so eine starke technische Unterstützung für das Proteindesign auf atomarer Ebene.
Versuchsergebnisse: La-Proteina führt in allen vier Tests mit großem Abstand
Um die Leistung von La-Proteina zu überprüfen, führte das Forschungsteam eine Reihe von Experimenten in zwei Hauptrichtungen durch: bedingungslose Proteinerzeugung auf atomarer Ebene und Gerüstdesign auf atomarer Motivebene, wobei die Leistung des Modells in verschiedenen Szenarien umfassend berücksichtigt wurde.
Im Experiment zur bedingungslosen Proteinproduktion im atomaren MaßstabWie in der Abbildung unten dargestellt, verglich das Forschungsteam zwei Varianten von La-Proteina (mit und ohne dreieckige Multiplikationsschichten) mit mehreren öffentlich verfügbaren All-Atom-Generation-Basismethoden wie P (All-Atom), APM und PLAID. Die Bewertungsindikatoren umfassten All-Atom-Kollaborationsdesignfähigkeiten, Diversität, Neuartigkeit und Standarddesignfähigkeiten.
Die Ergebnisse zeigen, dass die beiden Varianten von La-Proteina alle Basismethoden hinsichtlich der Fähigkeit zum All-Atom-Co-Design, der Designkapazität und der Vielfalt übertreffen und auch hinsichtlich der Neuartigkeit äußerst wettbewerbsfähig sind.
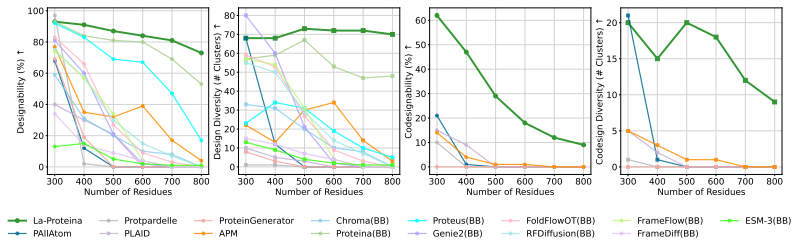
Die Fähigkeit von La-Proteina, unbedingt lange Ketten zu erzeugen
Es ist erwähnenswert, dass La-Proteina, das keine dreieckigen Multiplikationsschichten verwendet, eine hochmoderne Leistung bei hoher Skalierbarkeit erreicht, während das zweitbeste P (All-Atom) aufgrund seiner Abhängigkeit von rechenintensiven dreieckigen Aktualisierungsschichten nur kurze Proteine verarbeiten kann.
Auch,Das Forschungsteam demonstrierte außerdem die Skalierbarkeit von La-Proteina bei der Erzeugung großer rein atomarer Strukturen.Das Modell wurde anhand des AFDB-Datensatzes mit etwa 46 Millionen Proben trainiert und schneidet am besten bei der Generierung von Proteinen mit einer Länge von mehr als 500 Resten ab, während andere All-Atom-Baseline-Methoden häufig Schwierigkeiten haben, effektive Proben in diesem Längenbereich zu generieren.
Bei der biophysikalischen Analyse wurde das MolProbity-Tool zur Bewertung der Konstruktvalidität verwendet.Die Ergebnisse zeigten, dass die von La-Proteina erzeugte Struktur von höherer Qualität war.Das Ergebnis ist deutlich besser als bei allen Basismethoden, und die generierte Struktur ist auf physikalischer Ebene realistischer und ähnelt stärker dem realen Protein. Gleichzeitig wird durch die Visualisierung der Verteilung der Seitenketten-Diederwinkel und den Vergleich mit PDB- und AFDB-Referenzen festgestellt, dass, La-Proteina kann den Konformationsraum von Aminosäure-Rotationsisomeren genau simulieren,Die Basismethoden weichen häufig von der Referenz ab, es fehlen Muster oder es werden unrealistische Winkelbereiche ausgefüllt.
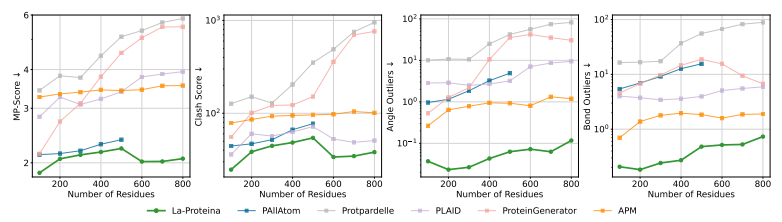
La-Proteina ist besser als die bestehende All-Atom-Generation-Baseline
Hat eine höhere Konstruktvalidität
Im Experiment zum Design des Atommotiv-GerüstsDas Forschungsteam bewertete die Leistung des Modells anhand der Aufgabe des atomaren Motivgerüstdesigns. Dabei muss das Modell eine Proteinstruktur generieren, die das Motiv basierend auf der Atomstruktur eines vordefinierten Motivs präzise unterstützt. Die Experimente wurden in vier Evaluierungsumgebungen durchgeführt, darunter All-Atom- und Advanced Atomic Scaffold Design sowie indizierte und nicht-indizierte Versionen.
Die Ergebnisse zeigen, dass in allen vier SettingsLa-Proteina übertrifft die einzige vergleichbare All-Atom-Baseline-Methode, Protpardelle, deutlich und ist in der Lage, die meisten Benchmark-Aufgaben erfolgreich zu lösen.Insbesondere bei Motiven, die aus drei oder mehr verschiedenen Restsegmenten bestehen, ist die Leistung der nicht indizierten Version von La-Proteina besser als die indizierte Version. Dies liegt wahrscheinlich daran, dass die Fixierung der Positionen mehrerer Segmente die Flexibilität des Modells bei der Untersuchung verschiedener Strukturlösungen einschränkt.
Wissenschaftliche Durchbrüche und innovative Verfahren im Bereich des Proteindesigns auf atomarer Ebene
Im Bereich des Proteindesigns hat die von La-Proteina vertretene Forschungsrichtung des Proteindesigns auf atomarer Ebene große Aufmerksamkeit in Wissenschaft und Wirtschaft erregt. Viele Universitäten und Unternehmen haben auf diesem Gebiet wichtige wissenschaftliche Durchbrüche und innovative Verfahren erzielt.
Im akademischen Bereich arbeiten einige Forschungsteams daran, die Leistung und Skalierbarkeit von Proteingenerierungsmodellen zu verbessern. So hat sich NVIDIA beispielsweise mit Mila, dem Quebec Institute for Artificial Intelligence, der Universität Montreal und dem Massachusetts Institute of Technology zusammengeschlossen, umDas entwickelte Proteina wurde auf der großen AlphaFold-Datenbank (AFDB) trainiert.Demonstrierte die Skalierbarkeit eines flussbasierten Modells zur Generierung von Proteinstrukturen.
Es gibt auch einige Studien, die Diffusionsmodelle im Proteindesign verwenden. Beispielsweise konzentrieren sich frühe diffusionsbasierte Proteingeneratoren wie RFDiffusion und Chroma auf die Proteinrückgrat-Generierung. Nachfolgende Studien haben den Anwendungsbereich von Diffusionsmodellen im Proteindesign weiter erweitert, beispielsweise durch Diffusion auf SO(3)-Mannigfaltigkeiten und euklidische Flussanpassungsmethoden.
Einige Forschungsteams konzentrieren sich auch auf die gemeinsame Modellierung von Proteinsequenz und -struktur. Beispielsweise nutzt der von NVIDIA und dem MIT eingeführte ProtComposer zusätzliche statistische Modelle und 3D-Primitive zur Generierung von Proteinstrukturen, während andere Arbeiten sich mit Allatomstrukturen befassen, indem sie Proteinrückgrat und -sequenz gemeinsam modellieren oder latente Variablenmodelle verwenden. Darüber hinaus wurden Sprachmodelle auch im Proteindesign eingesetzt, wobei sich einige Methoden auf Proteinsequenzen konzentrieren, während andere Strukturinformationen tokenisieren und Sequenzen und Strukturen gemeinsam modellieren.
In der Geschäftswelt konzentriert sich das niederländische Biotechnologieunternehmen Cradle auf den Einsatz künstlicher Intelligenz zur Vereinfachung des Proteindesignprozesses. Das Unternehmen hat ein Labor eingerichtet, in dem Milliarden von Proteinsequenzen und Daten gesammelt werden, um proprietäre generative KI-Modelle zu trainieren und so Proteindesign und -optimierung zu vereinfachen. Xaira Therapeutics, ein amerikanischer KI-Pharmadienstleister, entwickelt adaptive Moleküle für spezifische Indikationen und nutzt dabei seine Vorteile in der Forschung zu fortgeschrittenem maschinellem Lernen, der Generierung großer Datenmengen und der Therapieentwicklung. Einige Unternehmen kombinieren zudem Proteindesign-Technologie mit künstlicher Intelligenz und maschinellem Lernen, um die Effizienz und Genauigkeit des Proteindesigns zu verbessern.
Die wissenschaftlichen Forschungsdurchbrüche dieser Universitäten und die innovativen Praktiken der Unternehmen haben die Entwicklung des Proteindesigns mit reichhaltiger Erfahrung und technischer Unterstützung vorangetrieben und die kontinuierliche Weiterentwicklung dieses Bereichs gefördert. Mit dem kontinuierlichen technologischen Fortschritt wird Proteindesign künftig voraussichtlich in weiteren Bereichen eine wichtige Rolle spielen.
Referenzartikel:
1.https://mp.weixin.qq.com/s/7r69S3XpNMjemo3EiXzNeQ
2.https://mp.weixin.qq.com/s/DrZEdsb1SqSSkv_hbrp3TA








