Command Palette
Search for a command to run...
Durch Die Gleichzeitige Verarbeitung Von Informationen Zur Hauptkette Und Seitenkette Von Proteinen Gelang Stanford Et al. Die Modellierung Der Gesamten Atomstruktur Auf Der Grundlage Eines Neuronalen Nachrichtenübermittlungsnetzwerks.
Die Proteinseitenkettenkonformation beschreibt die spezifische räumliche Anordnung der Seitenketten von Aminosäureresten in Proteinen im dreidimensionalen Raum. Die Untersuchung der Proteinseitenkettenkonformation kann helfen, den Zusammenhang zwischen Proteinstruktur und -funktion zu verstehen und hat einen hohen Anwendungswert im Protein-Engineering, der Arzneimittelentwicklung und anderen Bereichen. Aktuelle Methoden des Proteinsequenzdesigns auf Basis von Deep Learning konzentrieren sich jedoch hauptsächlich auf das Design fixierter Hauptkettenproteinsequenzen und sind meist nicht in der Lage, die Proteinseitenkettenkonformation während der Sequenzgenerierung zu modellieren.Die wichtigsten Seitenketteninteraktionen werden nur auf Grundlage der Hauptkettengeometrie und bekannter Aminosäuresequenz-Tags abgeleitet, während die Rolle der Proteinseitenkettenkonformation in Proteinen ignoriert wird.
Um diese Lücke zu schließen, hat ein Team der Stanford University und des Arc Institute in Palo Alto, Kalifornien,Gemeinsam haben wir eine neue Methode zur Protein-Sequenz-Gestaltung vorgeschlagen, FAMPNN (Full-Atom MPNN), mit der die Sequenzidentität und Seitenkettenstruktur jedes Aminosäurerests explizit modelliert werden kann.Das Modell verwendet eine Nachrichtenübermittlungsarchitektur basierend auf Graph Neural Networks (GNNs), kombiniert mit verbesserten MPNN- (Message Passing Neural Networks) und GVP-Modulen (Geometric Vector Perceptron) für die All-Atom-Kodierung und kann gleichzeitig Informationen zur Proteinhauptkette und -seitenkette verarbeiten. Studien haben gezeigt, dass FAMPNN durch die explizite Modellierung von All-Atom-Strukturen die Qualität des Proteinsequenzdesigns und die Genauigkeit experimenteller Vorhersagen deutlich verbessern kann.
Das Forschungsergebnis mit dem Titel „Sidechain conditioning and modeling for full-atom protein sequence design with FAMPNN“ wurde für ICML 2025 ausgewählt.
Forschungshighlights:
* Wir führen eine Methode ein, die Kreuzentropie- und Diffusionsverlustziele kombiniert, um die Verteilung pro Label sowohl der diskreten Sequenzidentität von Resten als auch der kontinuierlichen Seitenkettenstruktur zu modellieren.
* Wir implementieren eine leichtgewichtige iterative Sampling-Methode zur Generierung von Samples aus einer gemeinsamen Verteilung und verwenden verbesserte MPNN- und GVP-Schichten für die All-Atom-Kodierung
* Untersuchungen haben gezeigt, dass FAMPNN die Genauigkeit des Sequenzdesigns und der experimentellen Proteinfitnessvorhersage durch die explizite Modellierung von All-Atom-Strukturen effektiv verbessern kann

Papieradresse:
Weitere Artikel zu den Grenzen der KI:
https://go.hyper.ai/owxf6
Datensätze: Vielfältige Datensätze optimieren das Training und die Auswertung von Modellen
Um die Wirksamkeit und Zuverlässigkeit des Modells sicherzustellen, verwendete das Forschungsteam komplexe Multi-Datasets für Training und Evaluierung:
In der Studie wurde hauptsächlich der S40-Datensatz von CATH 4.2 verwendet.Der Datensatz ist ein kuratierter Satz von Domänen, die aus der Protein Data Bank (PDB) extrahiert wurden, wobei redundante Domänen mit einer Homologie von mehr als 40% entfernt wurden, und der Datensatz im Verhältnis 8:1:1 in Trainings-, Validierungs- und Testsätze unterteilt wurde.
Der PDB-Datensatz basiert auf der gesamten PDB-Datenbank und umfasst Strukturen, die bis zum 30. September 2021 veröffentlicht wurden. Die Forscher gruppierten die Proteine entsprechend der Sequenzhomologie von 40% auf Proteinkettenebene und priorisierten Beispiele für mehrkettige Proteine für Trainingsmodelle, um das Entwerfen mehrkettiger Proteine zu erlernen.
Die Datensätze CASP13, 14 und 15 werden hauptsächlich verwendet, um die Leistung des Modells bei der Seitenkettenpackung zu bewerten.Das Forschungsteam verwendete die MMseqs2-Vorschlagssuche, um alle Sequenzen mit einer Homologie größer als 40% mit den CASP13-, 14- und 15-Datensätzen aus den Trainings- und Validierungsdatensätzen zu entfernen, und maß dann die Leistung der Seitenkettenpackung anhand der durchschnittlichen quadratischen Mittelabweichung (RMSD) zwischen den vorhergesagten Seitenketten und den tatsächlichen Seitenketten.
Der SKEMPlv2-Datensatz wurde verwendet, um die Vorhersagefähigkeit des Modells hinsichtlich der Protein-Protein-Bindungsaffinität zu bewerten.Der Datensatz fasst die experimentell gemessenen Bindungsaffinitäten von Tausenden von Sequenzvarianten in Hunderten von Protein-Protein-Interaktionen zusammen und nach der Verarbeitung umfasst der endgültige Datensatz 6.649 Datenpunkte.
Die Datensätze S669, Megascale und FireProtDB wurden verwendet, um die Zero-Shot-Vorhersagefähigkeit des Modells hinsichtlich der Proteinstabilität zu bewerten.Diese Datensätze enthalten experimentelle Messungen von Stabilitätsänderungen einer Vielzahl natürlicher Proteine (△△G), die häufig als Benchmark-Datensätze zur Stabilitätsvorhersage verwendet werden. Der Megascale-Datensatz ist eine ausgewählte, deduplizierte Version des Datensatzes. Das Forschungsteam hat Trainingssatz, Validierungssatz und Testsatz zu einem einzigen Datensatz zusammengeführt und so letztlich einen Datensatz mit 272.712 experimentellen Datenpunkten zu 298 verschiedenen Proteinen erhalten. Der FireProtDB-Datensatz enthält Änderungen der freien Energie von 3.438 Einzelmutationen von 100 einzigartigen Proteinen, von denen 3.420 Beispiele nach der Verarbeitung letztlich verwendet wurden. Der S669-Datensatz enthält experimentelle Messungen von 669 Einzelmutationen von 94 Proteinen, und 4 Varianten wurden aufgrund des Vorhandenseins nicht standardmäßiger Aminosäuren aus dem Datensatz ausgeschlossen.
Die Datensätze CR9114, CR6261 und G6 wurden verwendet, um die Leistung des Modells bei der Vorhersage der Antikörper-Antigen-Bindungsaffinität zu bewerten.Der Datensatz CR9114 enthält alle möglichen Kombinationen von 16 Aminosäuresubstitutionen. Der Datensatz CR6261 enthält alle möglichen Kombinationen von 11 Aminosäuresubstitutionen mit insgesamt 65.536 bzw. 2.048 Sequenzen. Der Datensatz G6 enthält insgesamt 4.275 Datenpunkte zur Bindung an VEGF-A.
Intelligentes Werkzeug zum gleichzeitigen Verständnis der Proteinsequenz und der Seitenkettenstruktur
Das Hauptziel dieser Studie besteht darin, dem Modell das gleichzeitige Erlernen der Proteinsequenz und der Seitenkettenkonformation zu ermöglichen. Zu diesem Zweck nutzte das Forschungsteam Masked Language Modeling, um FAMPNN basierend auf Sequenzkonsistenz zu trainieren.Das Training wird durchgängig durchgeführt, wobei kategorischer Kreuzentropieverlust (zur Sequenzvorhersage) und Diffusionsverlust (zur Vorhersage der Seitenkettenkonformation) kombiniert werden.Dadurch kann das Modell die maskierte Sequenz und ihre entsprechenden Seitenkettenkonformationen basierend auf teilweise bekannten Sequenzen und Seitenkettenkoordinaten gleichzeitig wiederherstellen.
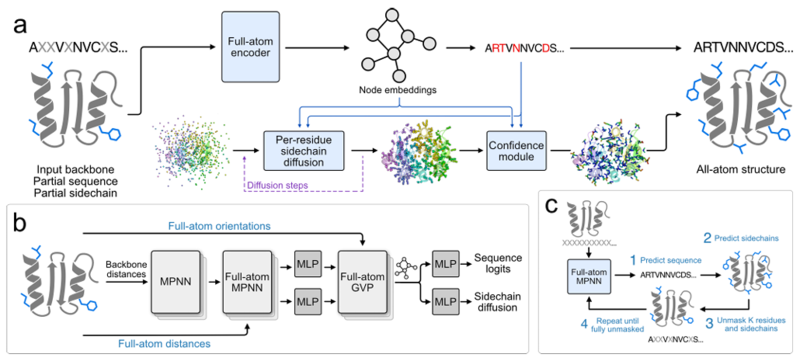
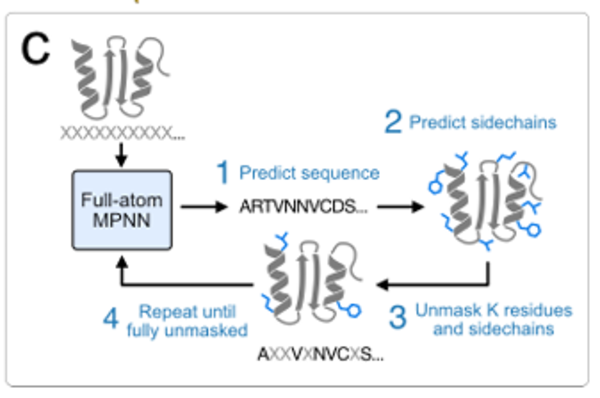
Beim Sampling wird eine iterative Sampling-Strategie ähnlich MaskGIT eingeführt. Dabei wird mit der vollständigen Maskierung von Sequenz und Seitenkette begonnen und die Maskierung einiger Sequenzen und Seitenketten-Token schrittweise vorhergesagt und entfernt, bis die vollständige Sequenz- und Seitenkettenstruktur vorliegt. Wie in der folgenden Abbildung dargestellt:
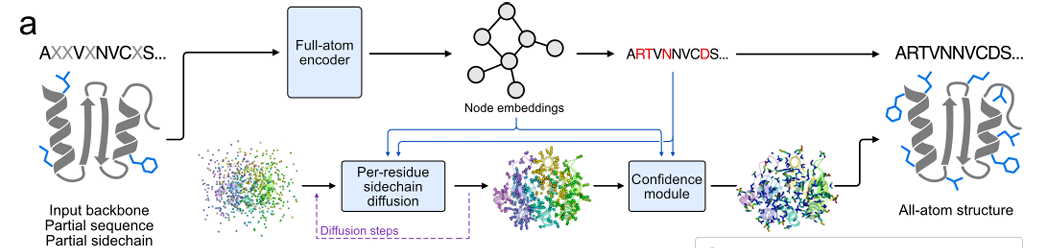
Im spezifischen Design werden die Seitenkettenkoordinaten im Atom37-Format dargestellt.Jeder Rest ist eine 37 x 3 Matrix mit fester Größe und 37 Atomen, einschließlich der 3D-Koordinaten von 4 Hauptkettenatomen (N, Cα, C und O) und 33 Seitenkettenatomen. Für Seitenketten ohne spezifischen Atomtyp werden Ghost-Atome (an der Cα-Position des Rests) verwendet, um sie darzustellen. Diese Methode löst das Problem der unterschiedlichen Anzahl von Seitenkettenatomen für verschiedene Aminosäuren.
Als Kern der Merkmalsextraktion verwendet der All-Atom-Encoder zur Kodierung ein Graph-Neuralnetzwerk mit hybrider MPNN-GVP-Architektur.Die Architektur besteht aus drei Hauptkomponenten: dem invarianten Hauptketten-Encoder, dem invarianten All-Atom-Encoder und dem äquivarianten All-Atom-Encoder. Die ersten beiden Komponenten basieren auf der Architektur von ProteinMPNN. Der invariante Hauptketten-Encoder ist identisch mit dem MPNN-Encoder, der nur die Hauptkettenstruktur kodiert; der invariante All-Atom-Encoder ersetzt den MPNN-Decoder, der identisch mit dem MPNN-Encoder des Hauptketten-Encoders ist, dessen Charakterisierung jedoch auf alle Atome erweitert ist. Die letzte Komponente verwendet ein verbessertes GVP, um dem Modell die Schlussfolgerung über vektorwertige interatomare Orientierungen zusätzlich zu den zuvor kodierten skalarwertigen interatomaren Abständen zu ermöglichen.
Zur Generierung der Seitenkettenkoordinaten verwendete das Forschungsteam die Methode der euklidischen Diffusion pro Token.Der Kern dieser Methode besteht in der Verwendung des euklidischen Diffusionsmodells (EDM) zur Lösung des Problems der Generierung kontinuierlicher Werte für Seitenketten-Atomkoordinaten. Ziel ist die Generierung einer Seitenkettenstruktur, die der Hauptkettenstruktur und der räumlichen Anordnung der umgebenden Aminosäuren entspricht. Während des Trainings wird den realen Seitenkettenkoordinaten zunächst zufälliges Rauschen hinzugefügt. Anschließend kann das Modell das Rauschen entfernen und die realen Koordinaten basierend auf dem Rauschpegel und bekannten Informationen wiederherstellen. Während der Inferenz entfernt das Modell ausgehend von den zufälligen Rauschkoordinaten schrittweise das Rauschen und generiert Seitenkettenkoordinaten, die den realen Koordinaten nahe kommen.
Um gleichzeitig den Einfluss der Gesamtrotation und -translation des Proteins auf die Seitenkettengenerierung zu vermeiden, werden die atomaren Koordinaten der Seitenkette während des Trainings in ein lokales Koordinatensystem basierend auf den Atomen der Hauptkette konvertiert und nach der Generierung wieder in das globale Koordinatensystem zurückkonvertiert.Die Eingabe des Diffusionsmodells umfasst die vom All-Atom-Encoder extrahierten Merkmale, die vorhergesagte Sequenzidentität und den aktuellen Rauschpegel.Die generierten Seitenkettenkoordinaten werden auch in der gemeinsamen Verlustfunktion verwendet, um das Modelltraining zu steuern, wie in der folgenden Abbildung dargestellt.
Um den Fehler der Modellvorhersage zu reduzieren und die Genauigkeit zu verbessern, entwickelte das Forschungsteam außerdem ein Konfidenzmodul zur Vorhersage von Sidechain-Packungsfehlern (Predicted Sidechain Error, pSCE). Konkret:Dieses Modul unterteilt den tatsächlichen Fehler der Seitenkettenatome (den Abstand zwischen den generierten Koordinaten und den wahren Koordinaten) in 33 Intervalle.Das Modell wird mit kategorialem Kreuzentropieverlust trainiert, um das Intervall jedes Atomfehlers basierend auf den Informationen im Generierungsprozess vorhersagen zu können. Anschließend wird die endgültige Fehlerschätzung pSCE durch die Erwartung der Intervallwahrscheinlichkeit ermittelt. Die Eingabe dieses Moduls umfasst die Eigenschaften des All-Atom-Encoders, die generierten Sequenz- und Seitenkettenkoordinaten sowie den Rauschpegel im Diffusionsprozess. Die Ausgabe pSCE kann die Genauigkeit der Seitenkettenpackung effektiv widerspiegeln, was hilfreich ist, um hochwertige Designergebnisse zu prüfen und die Interpretierbarkeit des Modells zu verbessern. Dadurch wird die Qualitätsbewertung der Seitenkettenstrukturgenerierung verbessert.
Experimentelle Ergebnisse: Die Leistung ist deutlich besser als bei dem Modell, das nur auf der Hauptkette basiert
Um die Leistung zu überprüfen und das Modell genau zu bewerten, führte das Forschungsteam zunächst Experimente zur Sequenzwiederherstellung und Selbstkonsistenzbewertung durch und verglich FAMPNN mit anderen Methoden. Die spezifischen Vergleichsobjekte sind in der folgenden Abbildung dargestellt.
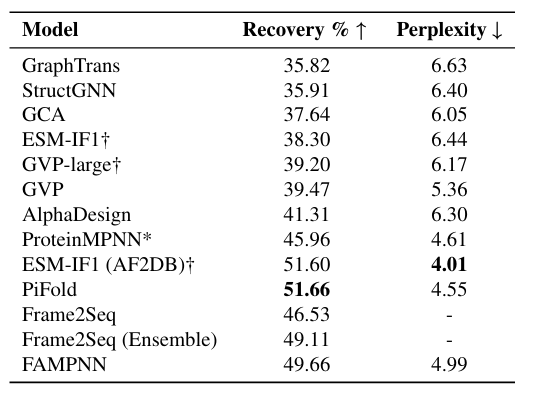
Experimente zeigen, dassFAMPNN übertrifft die aktuellen modernsten Methoden hinsichtlich der Genauigkeit der Einzelschritt-Sequenzwiederherstellung und erreicht 49,66%.Im Vergleich dazu beträgt ProteinMPNN nur 45,96% und GVP nur 39,47%.
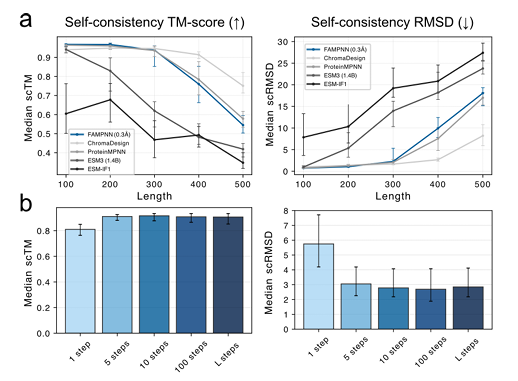
Bei der Selbstkonsistenzbewertung der neuen Hauptkette, die auf RFdiffusion basiert,FAMPNN (0,3 Å) ist hinsichtlich der scTM- (strukturelle Ähnlichkeit) und scRMSD-Metriken (root mean square deviation) mit ProteinMPNN vergleichbar.Und durch 10 Schritte iterativer Stichprobennahme lässt sich ein hohes Maß an Selbstkonsistenz erreichen, was effizienter ist als die vollständig autoregressive Methode. Wie in der folgenden Abbildung dargestellt:
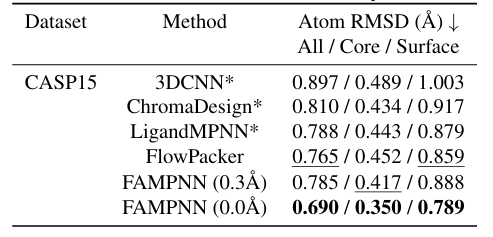
In Bezug auf die Sidechain-Packung verglichen die Forscher das vorgeschlagene Modell mit anderen Methoden anhand der Datensätze CASP13, 14 und 15.Experimente zeigen, dass bei der Kristallstrukturbewertung des CASP15-Testsatzes der atomare RMSD (All/Core/Surface) von FAMPNN (0,0 Å) 0,690/0,350/0,789 Å beträgt, was besser ist als bei anderen Methoden.Es besteht eine starke Korrelation mit dem Fehler jedes Atoms und dem Fehler jedes Rests, mit Spearman-Korrelationskoeffizienten von 0,843 bzw. 0,780. Wie in der folgenden Abbildung dargestellt:
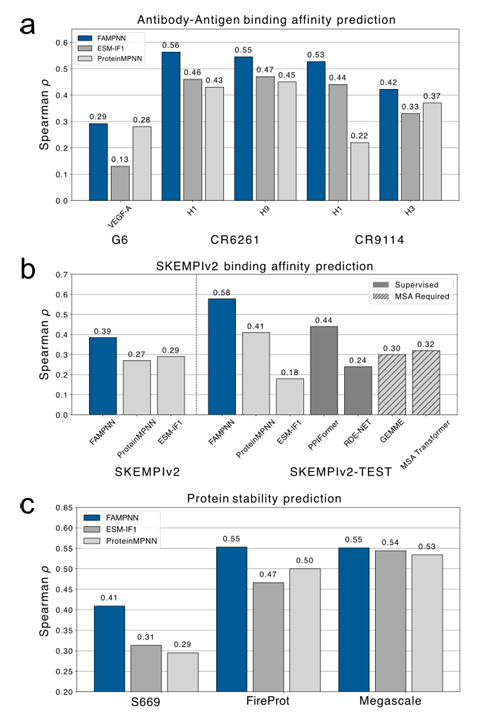
Bei der Beurteilung der Proteinfitness unter All-Atom-BedingungenIm SKEMPlv2-Datensatz übertraf FAMPNN das unüberwachte Modell deutlich und übertraf im Test-Subset sogar das überwachte Modell. Dies zeigte eine starke Generalisierungsfähigkeit bei der Nullstichprobenvorhersage. In den drei Stabilitätsdatensätzen S669, Megascale und FireProtDB schnitt FAMPNN etwas besser ab als ProteinMPNN und ESM-IF. Bei der Vorhersage der Antikörper-Antigen-Bindungsaffinität übertraf FAMPNN stets die fortschrittlichsten unüberwachten Methoden ProteinMPNN und ESM-IF1, was den Nutzen von FAMPNN für die Proteinstabilität und die Verbesserung der Protein-Protein-Interaktionen belegt. Wie in der folgenden Abbildung dargestellt:
In Experimenten, in denen untersucht wurde, ob All-Atom-Modellierung die Leistung des Sequenzdesigns verbessern kann,Die Studie ergab, dass das Hinzufügen von Seitenkettenpackungszielen und All-Atom-Bedingungseinstellungen die Sequenzgenauigkeit verbessern kann.Darüber hinaus verbessert sich die Leistung von FAMPNN und des Basismodells durch die Einspeisung weiterer Strukturinformationen. An der Protein-Protein-Schnittstelle ist die Modellierung von Seitenketteninteraktionen wichtiger. Die Bereitstellung eines partiellen Seitenkettenkontexts in Verbindung mit der Sequenz kann die Genauigkeit im Vergleich zur Bereitstellung eines nur partiellen Sequenzkontexts deutlich verbessern.
Darüber hinaus kann FAMPNN im Vergleich zu LigandMPNN den Seitenkettenkontext effektiver nutzen und Seitenkettenpackungen basierend auf unterschiedlichen Anzahlen von Partialsequenz- oder Seitenkettenkonformationskontexten durchführen. Je mehr Kontexte, desto höher die Packungsgenauigkeit. Wie in Abbildung c unten dargestellt:
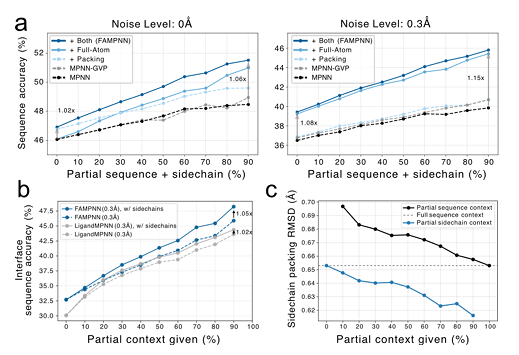
Zusammenfassend zeigen die obigen Experimente, dass FAMPNN im Vergleich zu Modellen, die nur auf der Hauptkette basieren, erhebliche Vorteile bei der Vorhersage der Proteinfitness hat.
Angetrieben von künstlicher Intelligenz floriert die Wissenschaft im Bereich der Sidechain-Modellierung
Wie eingangs erwähnt, ist die Seitenkettenkonformation entscheidend für die Proteinfunktion. Auch nach der Bestimmung der Hauptkette des Proteins gibt es jedoch noch viele mögliche Seitenkettenkonformationen, was die Modellierung und Erforschung von Seitenkettenkonformationen zu einer schwierigen Aufgabe macht. Zusätzlich zu dieser Studie treiben viele akademische Forschungseinrichtungen weltweit die Forschung zur Seitenkettenmodellierung mithilfe modernster Deep-Learning-Technologie und biologischem Wissen voran.
Ein Team der Fudan-Universität in China schlug eine zweistufige Methode zur Seitenkettenmodellierung namens OPUS-Rota5 vor.Diese Methode verwendet ein verbessertes 3D-Unet, um lokale Umgebungsmerkmale zu erfassen.Einschließlich Ligandeninformationen für jeden Rest und anschließender Verwendung des RotaFormer-Moduls zur Aggregation verschiedener Merkmalstypen. Auswertungen von Testsets wie CAMEO und CASP15 zeigen, dass OPUS-Rota5 einige andere führende Methoden zur Seitenkettenmodellierung deutlich übertrifft. Die entsprechende Forschung wurde auf ScienceDirect unter dem Titel „OPUS-Rota5: Eine hochpräzise Methode zur Proteinseitenkettenmodellierung mit 3D-Unet und RotaFormer“ veröffentlicht.
Papieradresse:
https://www.sciencedirect.com/science/article/pii/S0969212624001266
Ein Team der Peking-Universität schlug eine andere Methode namens GeoPacker vor.Diese Methode kombiniert geometrisches Deep Learning mit ResNet, um Proteinseitenketten zu modellieren. GeoPacker stellt atomare Wechselwirkungen rotations- und translationsinvarianter dar, um relative Positionsinformationen zu extrahieren. In Bezug auf die Vorhersagegenauigkeit der Seitenkettenstruktur übertrifft GeoPacker die fortschrittlichsten Methoden auf Basis von Energiefunktionen und läuft bei vergleichbarer Vorhersagegenauigkeit etwa 10- bzw. 700-mal schneller als die Deep-Learning-basierten Methoden DLPacker und OPUS-Rota4. Die zugehörige Forschung wurde unter dem Titel „GeoPacker: Ein neuartiges Deep-Learning-Framework für die Modellierung von Proteinseitenketten“ veröffentlicht.
Papieradresse:
https://onlinelibrary.wiley.com/doi/epdf/10.1002/pro.4484
Gleichzeitig schlug das Team der Universität Toronto ein Modell namens FlowPacker vor.Sein Zweck besteht darin, die spezifische Form der Seitenkette auf Grundlage der bekannten Aminosäuresequenz und Hauptkettenstruktur des Proteins genau vorherzusagen.Im Vergleich zu früheren fortschrittlichen Methoden schneidet FlowPacker bei den meisten Indikatoren besser ab und läuft schneller. Beispielsweise bietet es Vorteile bei Winkelvorhersagefehlern, der Nähe zwischen dem vorhergesagten Winkel und dem tatsächlichen Wert sowie der Abweichung der Atomposition usw. Die entsprechende Forschung wurde unter dem Titel „FlowPacker: Protein-Seitenkettenpackung mit Torsionsflussanpassung“ veröffentlicht.
Papieradresse:
Die Entschlüsselung von Seitenkettenkonformationen ist für die Entwicklung der Biowissenschaften von entscheidender Bedeutung. Die kontinuierliche Weiterentwicklung der künstlichen Intelligenz hat zweifellos die rasante Entwicklung der Strukturbiologie und der Computerbiologie vorangetrieben und in- und ausländischen Forschungseinrichtungen zu florierenden akademischen Leistungen verholfen. Sobald diese Erfolge vom Labor in die Anwendung umgesetzt werden, werden sie unweigerlich einen neuen Aufschwung in den Biowissenschaften auslösen und die Biowissenschaften und die Medizin in ein neues Kapitel führen.








