Command Palette
Search for a command to run...
Proteinstrukturvorhersage/Funktionsannotation/Interaktionserkennung/On-Demand-Design – Das Team Von Zhang Shugang Von Der China Ocean University Befasst Sich Direkt Mit Den Kernaufgaben Des Intelligenten Protein-Computings
Als Hauptträger des Lebens spielen Proteine eine Schlüsselrolle in den physiologischen Funktionen des Menschen. Die traditionelle Forschung steht jedoch vor Herausforderungen wie hohen Kosten für die Strukturanalyse, erheblichen Verzögerungen bei der funktionellen Annotation und geringer Effizienz bei der Entwicklung neuer Proteine. In den letzten Jahren ist die Analyse komplexer Proteineigenschaften in den Biowissenschaften immer dringlicher geworden. Die bahnbrechende Entwicklung von Technologien wie Big Data, Deep Learning und multimodalem Computing eröffnet neue Entwicklungsmöglichkeiten für den Aufbau eines proteinintelligenten Computersystems. Der Aufbau eines proteinintelligenten Computersystems ermöglichte es Proteinen, bemerkenswerte Ergebnisse in den Bereichen der groß angelegten funktionellen Annotation, der Interaktionsvorhersage und der dreidimensionalen Strukturmodellierung zu erzielen und eröffnete damit einen neuen technischen Weg für die Arzneimittelforschung und die Simulation von Lebenssystemen.
Auf der Beijing Zhiyuan Conference 2025 sprach Associate Professor Zhang Shugang von der School of Computer Science der Ocean University of China im Forum „AI+Science & Engineering & Medicine“ zum Thema „Konstruktion und Anwendung von proteinintelligenten Computersystemen“.Ausgehend vom Kernwert des intelligenten Protein-Computersystems erläutert das Dokument systematisch die technischen Durchbrüche von vier Kernaufgaben: Vorhersage der Proteinstruktur, funktionelle Annotation, Interaktionserkennung und neues Design.Die relevanten Forschungsergebnisse des Teams wurden hervorgehoben.

HyperAI hat die ausführlichen Ausführungen von Associate Professor Zhang Shugang zusammengestellt und zusammengefasst, ohne die ursprüngliche Absicht zu verletzen. Nachfolgend finden Sie die Abschrift der Rede.
Überblick über das proteinintelligente Computersystem: KI-getriebene Revolution in den Biowissenschaften
Die Bedeutung von Proteinen in der biowissenschaftlichen Forschung ist offensichtlich. Sie sind nicht nur ein Enzym, das biochemische Reaktionen katalysiert, sondern auch ein Botenstoff, der Signale überträgt, die strukturelle Grundlage des Körpers bildet und die „Waffe“ des Immunsystems zur Abwehr äußerer Feinde ist. Traditionelle Forschungsmethoden scheinen jedoch angesichts der komplexen Eigenschaften von Proteinen wirkungslos zu sein. Probleme wie die hohen Kosten der Strukturanalyse, erhebliche Verzögerungen bei der funktionellen Annotation und die geringe Erfolgsquote beim Proteindesign sind zu wichtigen Herausforderungen geworden.
Mit der Einführung der KI-Technologie hat sich diese Situation komplett umgekehrt. Im Jahr 2024 wurde der Nobelpreis für Chemie für Durchbrüche bei der Vorhersage und dem Design von Proteinstrukturen durch KI verliehen, was zweifellos einmal mehr die wichtige Rolle der KI in der Proteinforschung unterstrich.Proteinintelligentes Computing ermöglicht eine effiziente Simulation und Vorhersage komplexer Proteineigenschaften durch die Erstellung datengesteuerter Algorithmusmodelle.Darüber hinaus liefert es neue Ideen und Forschungsparadigmen zur Bewältigung der oben genannten Herausforderungen und eröffnet eine neue Ära für die Biowissenschaftsforschung.
Durchbruch bei der Kernaufgabe des Protein-Intelligent-Computing
Die Kernthemen des Protein Intelligent Computing sind die folgenden vier Kategorien:
Kann die Proteinstruktur von Grund auf vorhergesagt werden?
Vom Levinthal-Paradoxon zur Subversion von AlphaFold
Nehmen wir die Proteinfaltung als Beispiel: Ein Protein mit 100 Resten kann bis zu 10 mögliche Konformationen haben.200 Bei einer zufälligen Suche ist die benötigte Zeit viel länger als das Alter des Universums (13,8 Milliarden Jahre), was dem berühmten Levinthal-Paradoxon entspricht. Die tatsächliche Proteinfaltung kann jedoch innerhalb von Millisekunden bis Minuten abgeschlossen sein, was auf einen spezifischen Faltungspfad hindeutet.
Im Jahr 2018 versuchte die erste Generation des AlphaFold-Modells, das Problem mithilfe von Deep-Learning-Methoden zu lösen, indem das Residual-Convolution-Modul verwendet wurde, um den Abstand und den Torsionswinkel von Aminosäurepaaren vorherzusagen.Bei CASP13 war es den anderen Wettbewerbern mit großem Abstand voraus und sagte 25 Proteinstrukturen genau voraus.Der Zweitplatzierte hatte nur 3 richtig vorhergesagt.
Im Jahr 2021 erreichte das Modell der zweiten Generation einen qualitativen Sprung. AlphaFold2 verwendete HMMER und HH-Suite, um eine Mehrfachsequenzausrichtung und eine Vorlagensuche durchzuführen.Durch 48 Evoformer-Module und 8 Strukturmodule wird eine Proteinstrukturvorhersage mit atomarer Präzision erreicht.Eine Datenbank mit rund 214 Millionen Proteinmonomer-Vorhersagen wurde veröffentlicht. Der durchschnittliche Fehler zwischen der vorhergesagten Struktur und den Ergebnissen der Elektronenmikroskopie-Analyse überschreitet nicht eine Atombreite und erreicht damit den Standard „Hochpräzise“.
Im Jahr 2024 wird das Modell der dritten Generation die vollständige Vorhersage der Proteininteraktionsstruktur in vivo ermöglichen. AlphaFold3 hat einen qualitativen Sprung gemacht. Es kann nicht nur die Proteinstruktur vorhersagen,Darüber hinaus kann die Struktur von Komplexen aus Proteinen, Nukleinsäuren, kleinen Molekülen, Ionen und allen anderen Lebensmolekülen vorhergesagt werden.Es deckt fast alle Molekültypen in der PDB-Datenbank ab und bietet ein leistungsstarkes Tool zum Verständnis von Zellfunktionen und zur Behandlung von Krankheiten.
Können Proteinfunktionen automatisch annotiert werden: ein Durchbruch bei der Multi-Source-Datenfusion
Aufgrund der zukunftsweisenden Fortschritte von AlphaFold3 in der Proteinvorhersage hat unser Team beschlossen, den Forschungsschwerpunkt auf die Annotation von Proteinfunktionen und Interaktionsanalyse zu verlagern. Derzeit sind von den 250 Millionen Proteinsequenzen weltweit nur 0,51 TP3T mit einer präzisen funktionalen Annotation versehen. Das traditionelle Modell, das auf manueller Analyse durch BiologInnen beruht, war der Herausforderung massiver Datenmengen nicht gewachsen. Daher stellt der Einsatz von Deep Learning zur Annotation großer Mengen an Batches einen entscheidenden Durchbruch dar.
Unsere Erkundung in diesem Bereich begann im Jahr 2022. Wir zielen auf den Schwachpunkt der Branche ab, dass Strukturdaten der Elektronenmikroskopie, auf denen Deep Learning basiert, selten und kostspielig sind.Wir schlagen als Innovation vor, die von AlphaFold2 vorhergesagten virtuellen Strukturdaten beim Modelltraining zu verwenden.Diese Strategie, ähnlich der „Datenverbesserung“, erweiterte den Umfang der Trainingsdaten erheblich – von den fünf Millionen Proben, die herkömmliche Elektronenmikroskope liefern können, auf einen theoretisch großen Vorhersagedatenpool von Hunderten Millionen. Die experimentelle Überprüfung zeigt, dass das auf Basis von Vorhersagedaten trainierte Modell nicht nur die native Version übertrifft, sondern auch neue Proteinfunktionen entdecken kann, die mit herkömmlichen Methoden nicht identifiziert werden.
Titel des Artikels:Verbesserung der Vorhersageleistung von Proteinfunktionen durch Nutzung von AlphaFold-vorhergesagten Proteinstrukturen
Papieradresse:
https://pubs.acs.org/doi/10.1021/acs.jcim.2c00885
Im Hinblick auf technologische InnovationenUm das Problem des unzureichenden Informations-Minings zur Proteinstruktur zu lösen, hat unser Team eine Methode zur Vorhersage von Proteinfunktionen vorgeschlagen, die auf selbstüberwachter Graphenaufmerksamkeit basiert.Durch die Kodierung der Korrelationsinformationen von Resten innerhalb des Proteinmoleküls und die vollständige Nutzung der Distanzinformationen zwischen Resten als Hilfsaufgabe kann die Leistung der Proteinfunktionsvorhersage verbessert werden.
Titel des Artikels:SuperEdgeGO: Edge-Supervised Graph Representation Learning für eine verbesserte Vorhersage von Proteinfunktionen (bevorstehende Veröffentlichung)
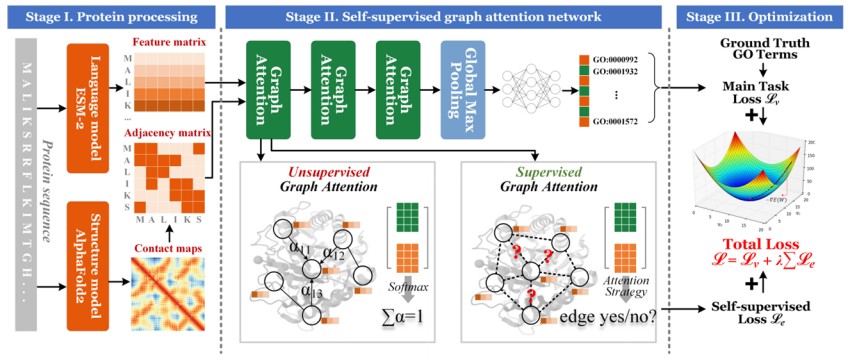
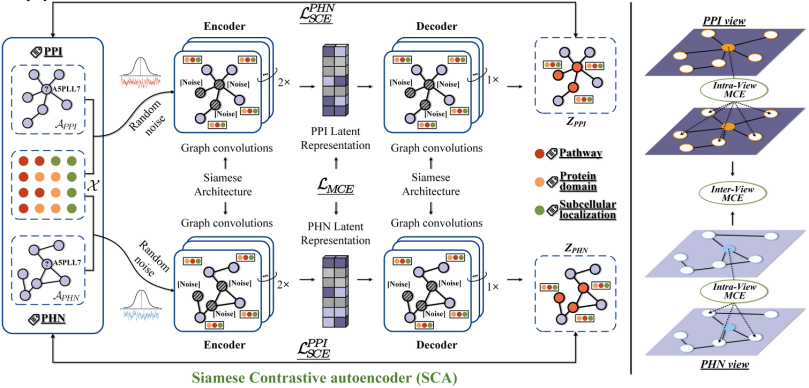
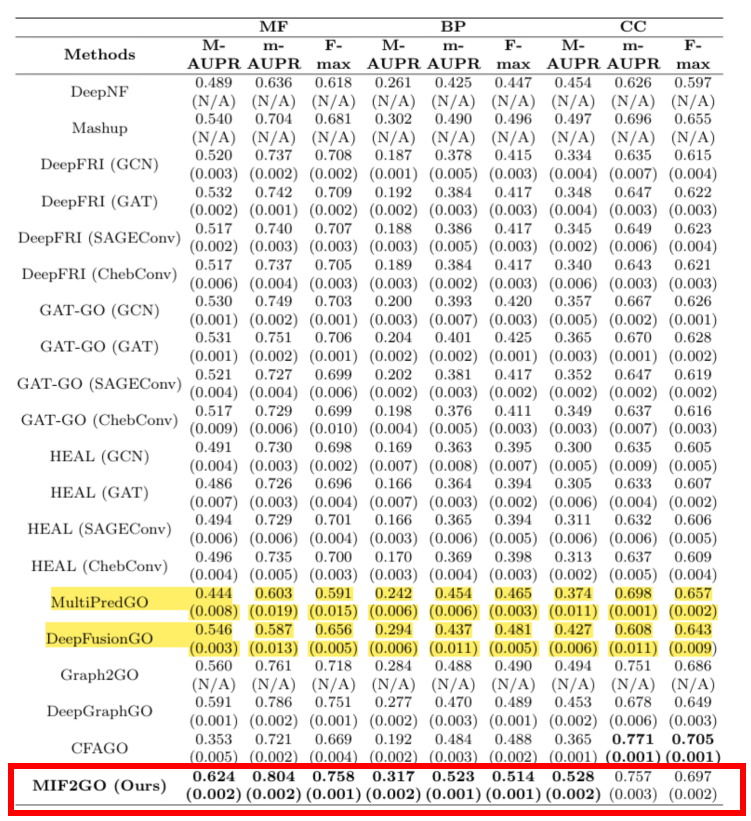
Um die Probleme heterogener Proteinmerkmale zu lösen, die schwer zu verschmelzen und räumlich inkonsistent sind, wurden eine Protein-Dual-View-Konstruktionsstrategie und eine Merkmalsausrichtungsmethode vorgeschlagen.Basierend auf den komplexen Eigenschaften biologischer Proteine mit sechs skalenübergreifenden Modi (die Dimensionen wie Sequenz, dreidimensionale Struktur und funktionelle Domäne abdecken),Das Team schlug außerdem eine multimodale Fusionsstrategie vor—— Integrieren Sie kontrastive Lern- und Multi-View-Analysemethoden im Computerbereich, um ein hierarchisches Merkmalsfusionsmodell zu erstellen. Diese Lösung wurde mit 20 gängigen Basismethoden anhand von 7 Datensätzen verglichen, die alle SOTA-Ergebnisse erzielten und das technische Problem der Leistungsverschlechterung durch direktes Spleißen von Modalitäten erfolgreich lösten.
Titel des Artikels:Annotation von Proteinfunktionen durch die Fusion mehrerer biologischer Modalitäten
Papieradresse:https://www.nature.com/articles/s42003-024-07411-y
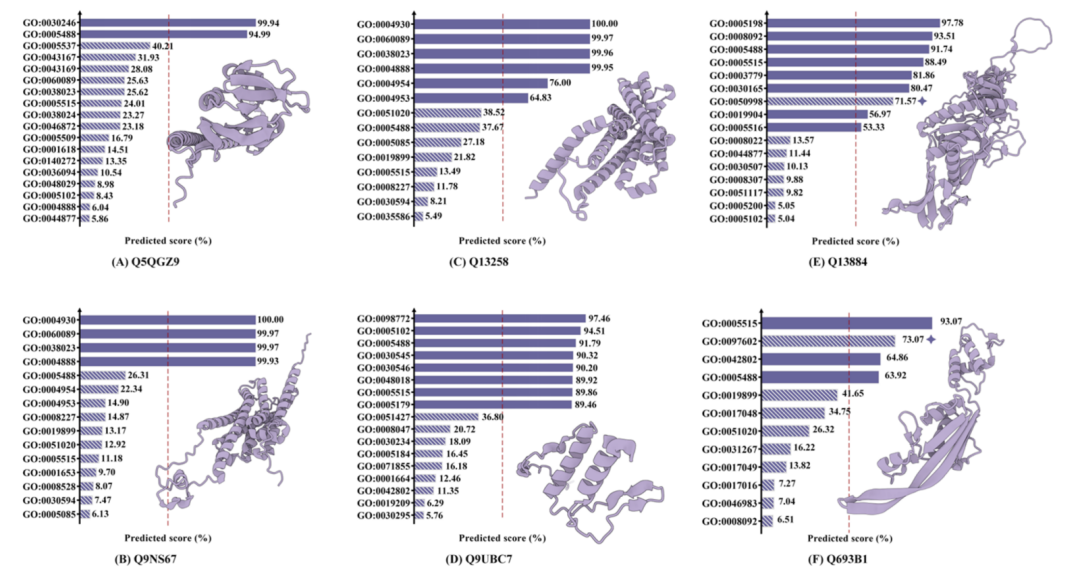
Darüber hinaus wurde bei der Untersuchung der Interpretierbarkeit funktionaler VorhersagenDas Modell zeigte außerdem eine hervorragende Fähigkeit, mehr als 10 Proteinfunktionen aus Tausenden von GoTerms-Anmerkungen genau zu identifizieren.Darüber hinaus fand das Team durch Literaturrecherche heraus, dass in einigen Studien Fälle auftraten, in denen das Modell Fehler vorhersagte, aber eine hohe Zuverlässigkeit bot. Dies deutet darauf hin, dass diese Fälle aufgrund der Verzögerung in der Datensatzversion möglicherweise falsch eingeschätzt wurden. Diese Entdeckung unterstreicht das Potenzial von KI-Modellen bei der Erforschung neuer Proteinfunktionen.
Lassen sich Proteininteraktionen präzise bestimmen? Selbst entwickelte Modelle ermöglichen effiziente Vorhersagen
In der Arzneimittelentwicklung ist das präzise Andocken von Proteinen als menschliche Zielmoleküle der Schlüssel zur Wirksamkeit von Medikamenten. KI-Technologie hat sich dabei als wertvoll erwiesen. Obwohl AlphaFold3 bei der Vorhersage von Proteinstrukturen gute Ergebnisse erzielt hat, gibt es in der praktischen Anwendung offensichtliche Einschränkungen: Die kostenlose Version unterstützt nur 20 Besuche pro Tag, deckt etwa 15 bis 20 Molekültypen ab und es ist äußerst schwierig, kommerzielle Nutzungsrechte zu beantragen. Dies veranlasste das Team, ein eigenes Modell zu entwickeln.
Ausgehend von dieser Problemstellung konzentrierte sich das Team auf die folgenden Aufgaben:
Zunächst haben wir uns mit den Problemen schwacher synergistischer Interaktionen bei bestehenden Methoden zur Vorhersage von Proteininteraktionen befasst.Im Encoder wird ein Zwillingslernmodell eingeführt, um die kollaborative Konsistenz der Proteindarstellung zu verbessern. Außerdem wird ein kollaborativer Lernrahmen mit einem kollaborativen Mechanismus für die Proteininteraktion und einem kollaborativen Mechanismus für Aufgaben vorgeschlagen.Das Team verwendete interaktive Aufmerksamkeits- und Multi-Task-Lernmethoden, um interaktive Vorhersagen von Protein-Nukleinsäure, Protein-Protein und Protein-kleinen Molekülen zu erreichen.
Das Team integrierte außerdem Transformer und Graph-Neuralnetzwerke in den NLP-Bereich und entwickelte Module wie Convformer und Graphormer, um eine interaktive Remote-Modellierung zu erreichen.Der Cross-Attention-Mechanismus wird genutzt, um die Fusion multimodaler Informationen zu verstärken. Das Modell zeigt in realen Szenarien eine hohe Generalisierungsfähigkeit. Am Beispiel der Vorhersage des Signalwegs bei Bauchspeicheldrüsenkrebs liegt die Genauigkeitsrate über 95%, wobei nur neun Paare von Interaktionsvorhersagefehlern auftreten.
Titel des Artikels:SSPPI: Modalitätsübergreifende verbesserte Vorhersage von Protein-Protein-Interaktionen aus Sequenz- und Strukturperspektive (bevorstehende Veröffentlichung)
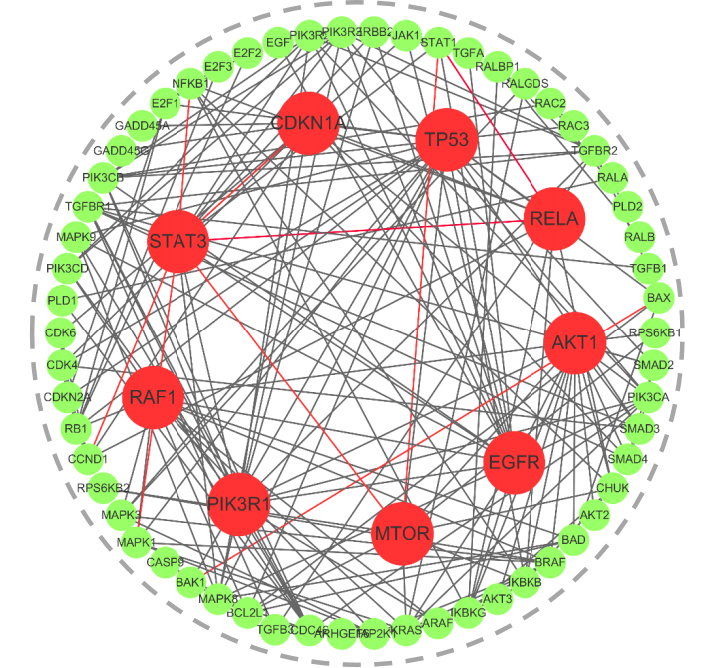
Schwarze Linie: richtige Vorhersage; rote Linie: falsche Vorhersage
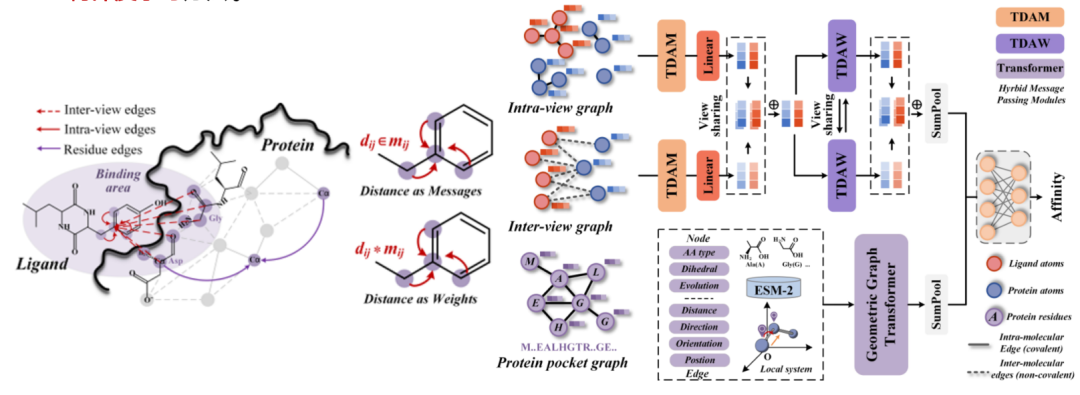
In unserer aktuellen Forschung beschäftigen wir uns neben der maßstabsübergreifenden Dimensionsreduktion von Proteinen auf Netzwerkebene auch mit der Gewinnung von Proteinmerkmalen. Da herkömmliche Graphenmodelle bei der Reduzierung dreidimensionaler Strukturinformationen auf zwei Dimensionen zu Informationsverlusten führen, haben wir das neueste geometrische Deep Learning eingeführt.Es wird eine geometrische Deep-Learning-Methode vorgeschlagen, die auf einer hybriden Nachrichtenübermittlungsstrategie basiert, und es wird ein vollständiges dreidimensionales Paradigma zur Informationsintegration erstellt.Dieses Paradigma zielt darauf ab, die Irrationalität des Verwerfens dreidimensionaler Informationen bei der räumlichen Standortmodellierung zu lösen und neue Forschungsideen im Bereich der dreidimensionalen Proteinmodellierung bereitzustellen.
Titel des Artikels:Geometrisches Deep Learning zur Vorhersage der Protein-Liganden-Affinität mit hybriden Nachrichtenübermittlungsstrategien (bevorstehende Veröffentlichung)
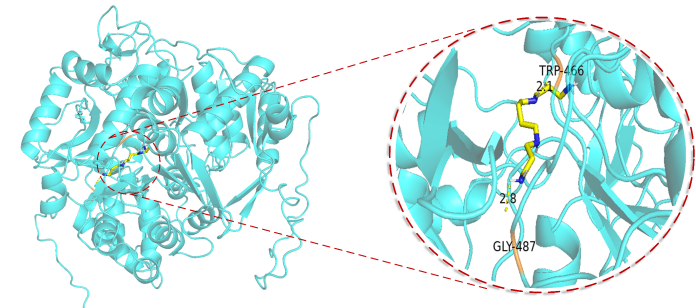
Auch,Wir haben auch tatsächliche Tests am ACSS2-Protein durchgeführt und mehrere Kandidatenverbindungen aus Zehntausenden von Verbindungen herausgesucht.Die Ergebnisse der Modellvorhersage deuten darauf hin, dass die Affinität der untersuchten Verbindungen den nM-Bereich erreichen kann, was auf ein gutes Arzneimittelpotenzial hindeutet. Unser Team arbeitete bei der Überprüfung mit dem Team des Qingdao University Medical College zusammen und die Docking-Ergebnisse wurden auch in den kürzlich durchgeführten Nassversuchen vorläufig bestätigt.
Können neue Proteine auf Anfrage entwickelt werden: Von inversen Problemen zu innovativen Anwendungen
Proteindesign ist eines der wichtigsten Ziele der Proteinforschung und von großer Bedeutung für die Impfstoffentwicklung, die Krebsbehandlung und die Entwicklung von Biomaterialien. Als inverses Problem der Proteinfaltung steht das Proteinsequenzdesign jedoch auch vor Herausforderungen wie der Suchraumexplosion und traditionellen Kraftfeldsimulationsfehlern.
Angesichts der Kernfrage des intelligenten Proteindesigns und der Proteinoptimierung betrachten wir hier die neueste Arbeit von Bakers Team, dem letztjährigen Nobelpreisträger, als Beispiel. Schlangengift besitzt kein spezifisches Gegenmittel. Ist es möglich, computergestützt ein neues Protein zu entwickeln? Ausgehend von dieser Problemstellung kombinierte Bakers Team seine bisherigen ProteinMPNN- und RFDiffusion-Verfahren, um ein neues Protein zu entwickeln. Darüber hinaus entwickelte sein Team spezifische Bindungsproteine für Schlangengifttoxine und lieferte damit eine neue Lösung zur Neutralisierung tödlicher Schlangengifttoxine. Die entsprechende Arbeit erschien Anfang 2025 im renommierten Fachjournal „Nature“. Diese Forschungsergebnisse zeigen das enorme Potenzial der KI im Bereich des Proteindesigns und sind ein wichtiger Schritt auf dem Weg zum kreativen Ziel, neue Proteine zu entwickeln.
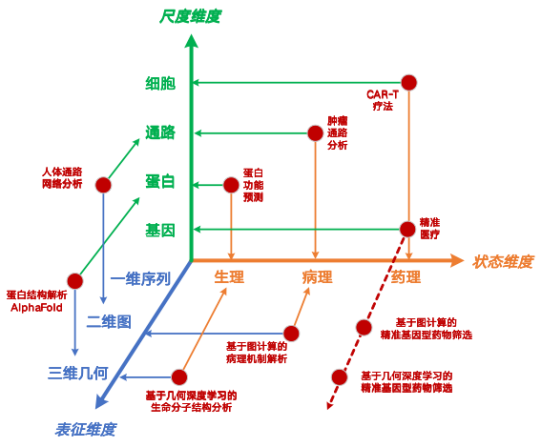
Skalenübergreifendes Computing komplexer Lebenssysteme: Simulation der gesamten Kette vom Nano- bis zum Makromaßstab
Das Lebenssystem ist ein komplexes, vielschichtiges System. Von der nanoskopischen Genebene bis zur makroskopischen Zellebene interagieren und beeinflussen sich alle Ebenen gegenseitig. Während meines Besuchs in der Forschungsgruppe von Professor Zhang Henggui an der Universität Manchester, Großbritannien, forschte ich zum digitalen Herzen. Nach meiner Rückkehr nach China forschte ich weiter an digitalen Zellen. Anders als beim digitalen Herzen, das auf numerischen Antrieben basiert,Das Team schlug eine mehrskalige Modellierungsmethode für mikroskopische Lebensaktivitäten auf der Grundlage eines „datengesteuerten“ Konstruktionsansatzes vor und konstruierte ein dreidimensionales mikroskopisches Rechenmethodensystem mit „Darstellung-Zustand-Skala“.Es deckt 36 Forschungspunkte ab und derzeit liegen zu fast einem Drittel der Methoden Artikel oder Patente vor.
Darüber hinaus unter der Leitung von Professor Wei Zhiqiang,Wir haben das mikroskopische Lebenssystem auf vier Maßstabsebenen neu definiert.Unter Einbeziehung der nanoskopischen Genebene, der „mikroskopischen“ Proteinebene, der „mesoskopischen“ Signalwegebene und der „makroskopischen“ Zellebene wird die Simulation des gesamten Lebenssystems durchgeführt, in der Hoffnung, eine vollständige Kopplung von den Atomen bis zum Herzen zu erreichen.
Über Associate Professor Zhang Shugang
Zhang Shugang ist außerordentlicher Professor und Master-Betreuer an der School of Computer Science der Ocean University of China, hochrangiges Mitglied des CCF, korrespondierendes Mitglied des CCF Bioinformatics Committee, Mitglied des CAAI Smart Healthcare Committee, Direktor der Shandong Bioinformatics Society und Direktor der National Natural Science Foundation of China, des Basic Scientific Research Business Expenses Project of Central Universities usw. Er wurde für das Shandong Postdoctoral Innovation Talent Support Program 2020 ausgewählt.
Seine Hauptforschungsgebiete sind Computerbiologie und Bioinformatik, einschließlich der Konstruktion digitaler Herzen mit ultrahoher Präzision, der Vorhersage und Gestaltung von Proteinfunktionen usw. In den letzten Jahren hat er über 30 Artikel in international anerkannten Fachzeitschriften und Konferenzen wie IEEE JBHI, JCIM und npj Systems Biology and Applications veröffentlicht, mit über 1.600 Zitierungen bei Google Scholar.








