Command Palette
Search for a command to run...
8k Lange Sequenzmodellierung, Proteinsprachenmodell Prot42 Kann Hochaffine Binder Nur Unter Verwendung Der Zielproteinsequenz Erzeugen
Proteinbinder (wie Antikörper und inhibitorische Peptide) spielen in wichtigen Szenarien wie der Krankheitsdiagnose, der Bildanalyse und der gezielten Arzneimittelverabreichung eine unersetzliche Rolle.Traditionell stützte sich die Entwicklung hochspezifischer Proteinbinder stark auf experimentelle Techniken wie Phagen-Display und gerichtete Evolution.Allerdings sind derartige Methoden im Allgemeinen mit der Herausforderung eines enormen Ressourcenverbrauchs und langer Forschungs- und Entwicklungszyklen konfrontiert und werden durch den inhärenten Engpass der Komplexität von Proteinsequenzkombinationen eingeschränkt.
Mit der Entwicklung künstlicher Intelligenz sind Protein-Sprachmodelle (PLMs) zu einem wichtigen Werkzeug für das Verständnis der Beziehung zwischen Proteinsequenzen und -funktionen geworden. Für das Design von Proteinbindern können PLMs dank der Generierungsfähigkeit von Sprachmodellen Ligandenproteine oder Antikörperfragmente mit hoher Bindungsaffinität direkt auf der Basis der Zielproteinsequenz entwickeln. Allerdings gibt es auch Herausforderungen, wie beispielsweise den Mangel an PLMs mit Fähigkeiten zur Langkontextmodellierung und echter Generierung, insbesondere beim Design komplexer Bindungsschnittstellen und langer Proteinbinder. Es besteht eine erhebliche technische Lücke.
Auf dieser Grundlage hat ein gemeinsames Forschungsteam des Inception AI Institute in Abu Dhabi, VAE und Cerebras Systems im Silicon Valley, USA,Es wurde die erste PLM-Familie, Prot42, vorgeschlagen, die nur auf Proteinsequenzinformationen beruht und keine dreidimensionale Struktureingabe erfordert.Dieses Modell nutzt die generative Kraft der Autoregression und der Nur-Decoder-Architektur.Ermöglicht die Erzeugung von Proteinbindern mit hoher Affinität und sequenzspezifischen DNA-bindenden Proteinen ohne Strukturinformationen.Prot42 zeigte im PEER-Benchmark, bei der Proteinbindergenerierung und bei DNA-sequenzspezifischen Bindergenerierungsexperimenten gute Ergebnisse.
Die zugehörige Forschung trägt den Titel „Prot42: eine neuartige Familie von Proteinsprachenmodellen für die zielbewusste Generierung von Proteinbindern“ und wurde als Vorabdruck auf arXiv veröffentlicht.
Forschungshighlights* Prot42 verwendet eine progressive Kontexterweiterungs-Trainingsstrategie, die schrittweise von anfänglich 1.024 Aminosäuren auf 8.192 Aminosäuren erweitert wird. * Im PEER-Benchmark-Test schneidet Prot42 bei 14 Aufgaben gut ab, darunter bei der Vorhersage von Proteinfunktionen, der subzellulären Lokalisierung und der Interaktionsmodellierung. * Im Gegensatz zu AlphaProteo, das auf 3D-Strukturen basiert, benötigt Prot42 lediglich die Zielproteinsequenz zur Generierung von Bindern.
Papieradresse:
Weitere Artikel zu den Grenzen der KI:
https://go.hyper.ai/UuE1o
Das Open-Source-Projekt „awesome-ai4s“ vereint mehr als 100 AI4S-Papierinterpretationen und stellt umfangreiche Datensätze und Tools bereit:
https://github.com/hyperai/awesome-ai4s
Datensätze: 3 große Datensätze unterstützen die Modellentwicklung und das Training
In dieser Studie wurden mehrere wichtige Datensätze verwendet, um das Modell zu trainieren und seine Leistung zu bewerten. Diese Datensätze decken nicht nur ein breites Spektrum an Proteinsequenzinformationen ab, sondern beinhalten auch Daten zur Protein-DNA-Interaktion und bieten somit umfangreiches Trainingsmaterial für Prot42.
Protein-DNA-Schnittstellendatenbank (PDIdb) 2010
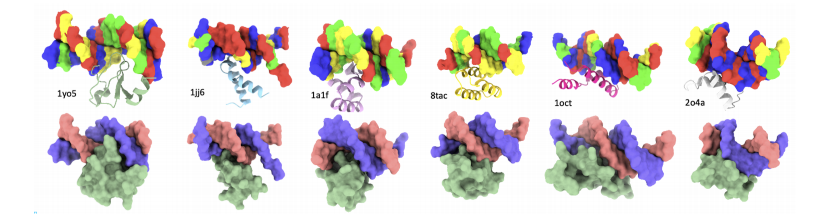
Um Proteine zu entwickeln, die an Ziel-DNA-Sequenzen binden können, verwendeten die Forscher den PDIdb 2010-Datensatz.Dieser Datensatz enthält 922 einzigartige DNA-Protein-Paare und wurde verwendet, um die Fähigkeit von Prot42 zu trainieren und zu bewerten, Proteine zu erzeugen, die an bestimmte DNA-Sequenzen binden.Um die vier DNA-Protein-Modelle zu bewerten, extrahierten die Forscher DNA-Fragmente aus verschiedenen PDB-Strukturen, darunter 1TUP, 1BC8, 1YO5, 1L3L, 2O4A, 1OCT, 1A1F und 1JJ6.
UniRef50-Datensatz
Der Vortrainingsdatensatz des Prot42-Modells stammt hauptsächlich aus der UniRef50-Datenbank.Die Datenbank enthält 63,2 Millionen Aminosäuresequenzen und deckt ein breites Spektrum biologischer Arten und Proteinfunktionen ab.Diese Sequenzen werden gruppiert und Sequenzen mit einer Ähnlichkeit von mehr als 50% werden zusammengefasst, wodurch die Datenredundanz reduziert und die Trainingseffizienz verbessert wird.
Vor dem Training von Prot42 hat das Forschungsteam den UniRef50-Datensatz vorverarbeitet.Sie werden mit einem Vokabular von 20 Standardaminosäuren beschriftet.Verwenden Sie Xtoken, um Aminosäurereste darzustellen (X wird verwendet, um ungewöhnliche oder mehrdeutige Aminosäurereste zu markieren).
In der Phase der DatenvorverarbeitungDas Forschungsteam verarbeitete die Sequenzen mit einer maximalen Kontextlänge von 1.024 Token und schloss längere Sequenzen aus, wodurch schließlich ein gefilterter Datensatz von 57,1 Millionen Sequenzen entstand.Die anfängliche Fülldichte beträgt 27%. Um die Datennutzung und Rechenleistung zu verbessern, hat das Forschungsteam eine Füllstrategie mit variabler Sequenzlänge (VSL) übernommen.Wir haben die Belegungsrate der Token innerhalb einer festen Kontextlänge maximiert und den Datensatz schließlich auf 16,2 Millionen aufgefüllte Sequenzen reduziert.Die Füllleistung erreicht 96%.
STRING-Datenbank
Die STRING-Datenbank ist eine umfassende Protein-Protein-Interaktionsdatenbank.Es integriert experimentelle Daten, rechnergestützte Vorhersagen und Text-Mining-Ergebnisse, um Konfidenzwerte für Proteininteraktionen zu ermitteln. Um Prot42 für die Generierung von Proteinbindern zu trainieren, filterte das Forschungsteam Proteininteraktionspaare mit Konfidenzwerten ≥ 90% aus der STRING-Datenbank, um die hohe Zuverlässigkeit der Trainingsdaten sicherzustellen.Darüber hinaus wurde die Sequenzlänge auf 250 Aminosäuren begrenzt, um sich auf handhabbare Einzeldomänen-Bindungsproteine zu konzentrieren.Nach dem Screening enthält der endgültige Datensatz 74.066 Protein-Protein-Interaktionspaare, einen Trainingssatz D(train)(pb) mit 59.252 Proben und einen Validierungssatz D(val)(pb) mit 14.814 Proben.
Modellarchitektur: 2 Hauptvarianten, abgeleitet von der autoregressiven Decoderarchitektur
Das in diesem Artikel erwähnte Prot42 ist ein PLM, das auf einer autoregressiven Decoderarchitektur basiert. Es generiert Aminosäuresequenzen nacheinander und sagt die nächste Aminosäure anhand der zuvor generierten Aminosäure voraus. Diese Architektur ermöglicht es dem Modell, Fernabhängigkeiten in der Sequenz zu erfassen.Es ist in der Lage, umfangreiche Darstellungen direkt aus großen, unmarkierten Proteinsequenzdatenbanken zu lernen und so die Lücke zwischen der riesigen Anzahl bekannter Proteinsequenzen und dem relativ geringen Anteil an Proteinsequenzen (<0,3%) effektiv zu schließen.Gleichzeitig enthält das Modell mehrere Transformer-Ebenen, von denen jede einen mehrköpfigen Self-Attention-Mechanismus und ein Feedforward-Neuralnetzwerk enthält, um komplexe Muster in der Sequenz zu erfassen.
Sein Design ist von Durchbrüchen in der natürlichen Sprachverarbeitung inspiriert, insbesondere vom LLaMA-Modell. Prot42 erfasst die evolutionären, strukturellen und funktionellen Informationen von Proteinen durch Vortraining an umfangreichen, unmarkierten Proteinsequenzen und ermöglicht so die Erzeugung von Proteinbindern mit hoher Affinität.
Auf dieser GrundlageDie Forscher trainierten zwei Modellvarianten vorab,Das sind Prot42-B und Prot42-L.
* Prot42-B:In der Basisversion verfügt das Modell über 500 Millionen Parameter und unterstützt eine maximale Sequenzlänge von 1.024 Aminosäuren.
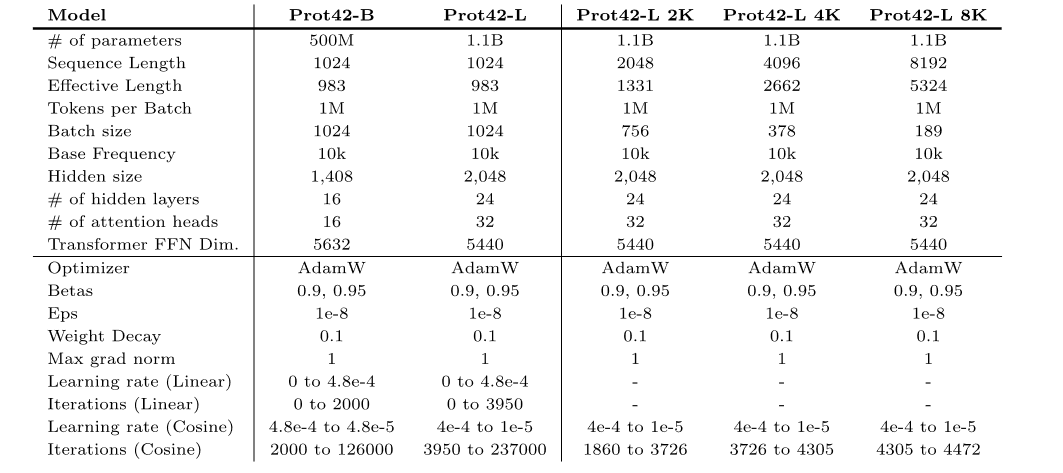
* Prot42-L:Die große Version verfügt über 1,1 Milliarden Modellparameter und unterstützt zudem eine maximale Sequenzlänge von 1.024 Aminosäuren.Die Kontextlänge von Prot42-L wurde schrittweise von 1.024 Aminosäuren auf 8.192 Aminosäuren erweitert.Dabei wurden eine schrittweise zunehmende Kontextlänge und eine konstante Batchgröße (1 Million nicht ausgefüllte Token) verwendet, um die Stabilität und Effizienz des Modells bei der Verarbeitung langer Sequenzen sicherzustellen und so die Fähigkeit des Modells zur Verarbeitung langer Sequenzen und komplexer Proteinstrukturen deutlich zu verbessern.Prot42-L enthält außerdem 24 versteckte Schichten mit jeweils 32 Aufmerksamkeitsköpfen.Die Dimension der verborgenen Schicht beträgt 2.048.
Experimentelles Fazit: Großes Potenzial zeigt sich in allen 6 Aufgaben
Um die Leistung des Prot42-Modells vor der Validierung anhand nachgelagerter Aufgaben zu bewerten, verwendeten die Forscher das Standardmaß der parametrischen Komplexität (PPL) zur Bewertung autoregressiver Sprachmodelle, nämlich die Leistung des Prot42-Modells bei unterschiedlichen Kontextlängen.Alle Modelle weisen bei 1.024 Token eine relativ hohe Perplexität auf, verbessern sich jedoch deutlich auf etwa 6,5 bei 2.048 Token.Die Ergebnisse zeigen, dass das Basismodell und die für kürzere Kontexte optimierten Modelle über ihre jeweiligen maximalen Kontextlängen hinweg ähnliche Leistungsmuster aufweisen. Besonders bemerkenswert ist die Leistung des 8k-Kontextmodells: Obwohl seine Perplexität bei Sequenzen mittlerer Länge (2.048 – 4.096 Token) etwas höher ist, kann es Sequenzen bis zu 8.192 Token verarbeiten und erreicht bei maximaler Länge eine minimale Perplexität von 5,1.Nach mehr als 4.096 Token zeigt die Perplexitätskurve einen Abwärtstrend.Wie in der Abbildung unten gezeigt.
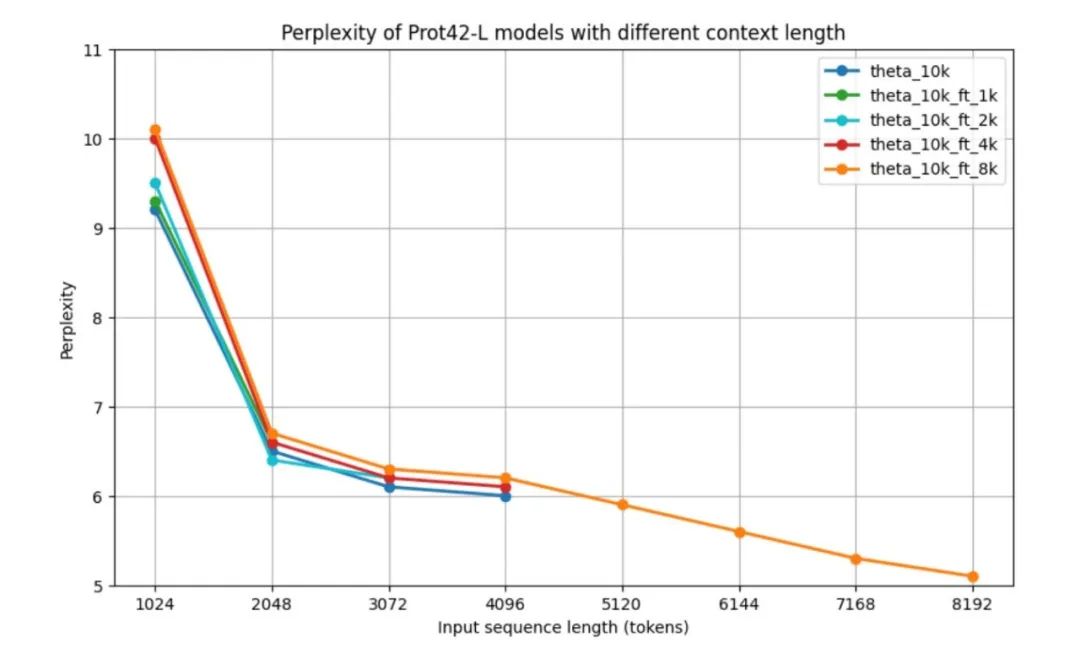
Mit zunehmender Kontextlänge nimmt der PPL des Modells allmählich ab, was darauf hinweist, dass die Fähigkeit des Modells, lange Sequenzen zu verarbeiten, erheblich verbessert wurde.Insbesondere erreicht das 8K-Kontextmodell den niedrigsten PPL, was darauf hindeutet, dass es das erweiterte Kontextfenster effektiv nutzen kann, um weitreichende Abhängigkeiten in Proteinsequenzen zu erfassen.Das erweiterte Kontextfenster stellt einen bedeutenden Fortschritt auf dem Gebiet der Protein-Sequenzmodellierung dar und ermöglicht eine genauere Darstellung komplexer Proteine und Protein-Protein-Interaktionen, was für die Erzeugung wirksamer Proteinbinder von entscheidender Bedeutung ist.
Durch eine Reihe strenger experimenteller BewertungenProt42 hat bei mehreren wichtigen Aufgaben eine hervorragende Leistung gezeigt.Es hat sich als wirksam bei der Erzeugung von Proteinbindern und der Entwicklung von Proteinen erwiesen, die an spezifische DNA-Sequenzen binden.
Vorhersage der Proteinfunktion
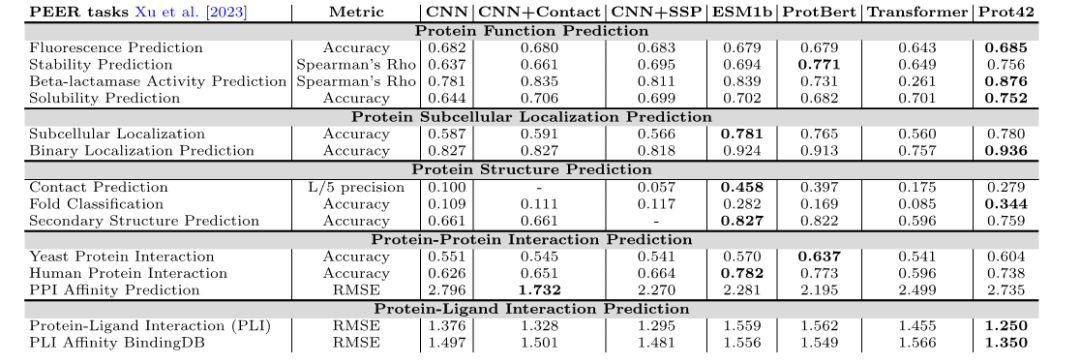
Im PEER-Benchmark-Test zeigte das Prot42-Modell gute Ergebnisse bei der Vorhersage mehrerer Proteinfunktionen, darunter Fluoreszenzvorhersage, Stabilitätsvorhersage, Vorhersage der β-Lactamase-Aktivität und Löslichkeitsvorhersage. Im Vergleich zu bestehenden ModellenProt42 hat erhebliche Vorteile bei der Vorhersage der Stabilität, Löslichkeit und β-Lactamase-Aktivität erzielt.Dies weist auf sein großes Potenzial bei hochauflösenden Protein-Engineering-Aufgaben hin.
Vorhersage der subzellulären Proteinlokalisierung
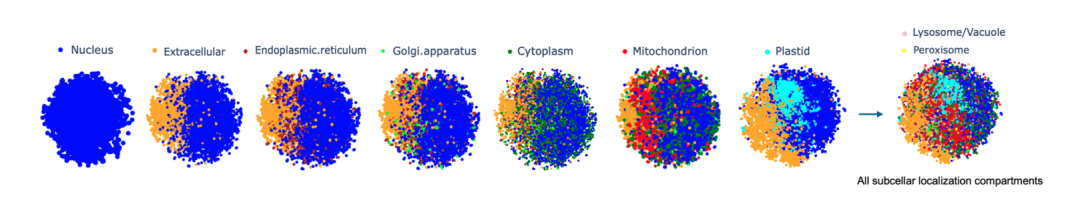
Die Forscher stellten jede Proteinsequenz als hochdimensionalen Vektor der Größe 32×2048 dar, betteten das Prot42-L-Modell in die gesamte Proteinsequenz ein und führten Berechnungen durch. Um die Qualitätsdifferenzierung in Einbettungen und Kompartimenten intuitiv bewerten zu können, verwendeten sie die t-verteilte stochastische Nachbareinbettung (t-SNE), um die Dimensionalität zu reduzieren und die Visualisierung von Proteingruppen übersichtlich zu gestalten.Es wurde nachgewiesen, dass Prot42 bei der Vorhersage der subzellulären Proteinlokalisierung gute Ergebnisse liefert und dass seine Genauigkeit mit der bestehender fortgeschrittener Modelle vergleichbar ist.Durch visuelle Analyse überprüfte das Forschungsteam außerdem die Wirksamkeit des Prot42-Modells bei der Erfassung der subzellulären Lokalisierungsmerkmale von Proteinen.
Vorhersage der Proteinstruktur
Bei der Aufgabe zur Vorhersage der ProteinstrukturDas Prot42-Modell erzielte hervorragende Ergebnisse bei der Kontaktvorhersage, Faltungsklassifizierung und Sekundärstrukturvorhersage.Diese Ergebnisse zeigen, dass das Prot42-Modell in der Lage ist, subtile Unterschiede in der Proteinstruktur zu erfassen und so eine starke Unterstützung für die Modellierung komplexer biologischer Interaktionen und pharmazeutischer Anwendungen darstellt.
Vorhersage von Protein-Protein-Interaktionen
Bei den Vorhersageaufgaben der Protein-Protein-Interaktion und der Protein-Liganden-Interaktion zeigte das Prot42-Modell eine hohe Genauigkeit und Zuverlässigkeit.Die Forscher verwendeten Chem42, um chemische Einbettungsvektoren zu generieren, und verglichen sie mit ChemBert., als weiteres chemisches Darstellungsmodell, sind seine Leistungsindikatoren dennoch besser als die bestehender Methoden und kommen den mit Chem42 erzielten Ergebnissen nahe. Insbesondere bei der Verwendung von Chem42 zur Generierung chemischer Einbettungen liegen die Vorhersageergebnisse nahe an denen professioneller chemischer Modelle.Dies deutet darauf hin, dass Prot42 über eine gute Erweiterbarkeit bei der Kombination chemischer Informationen verfügt.Bietet starke Unterstützung für die Arzneimittelentwicklung.
Proteinbinder-Generierung
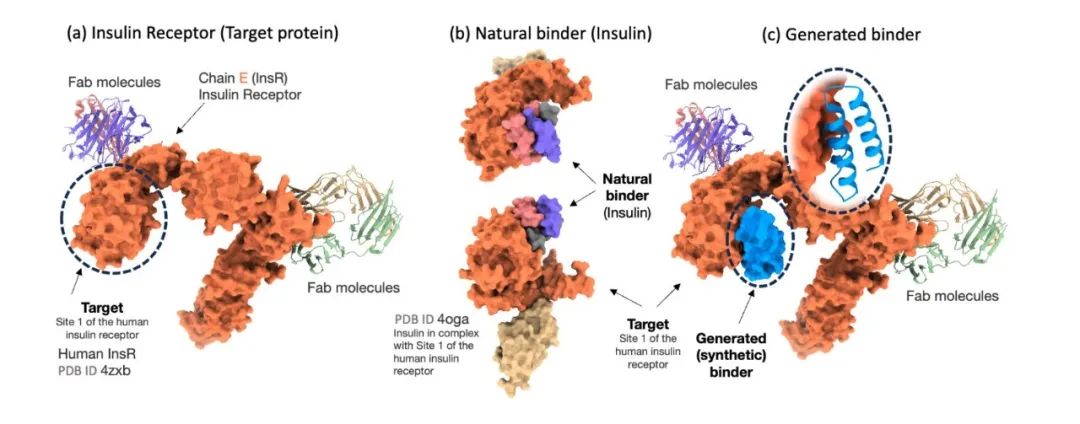
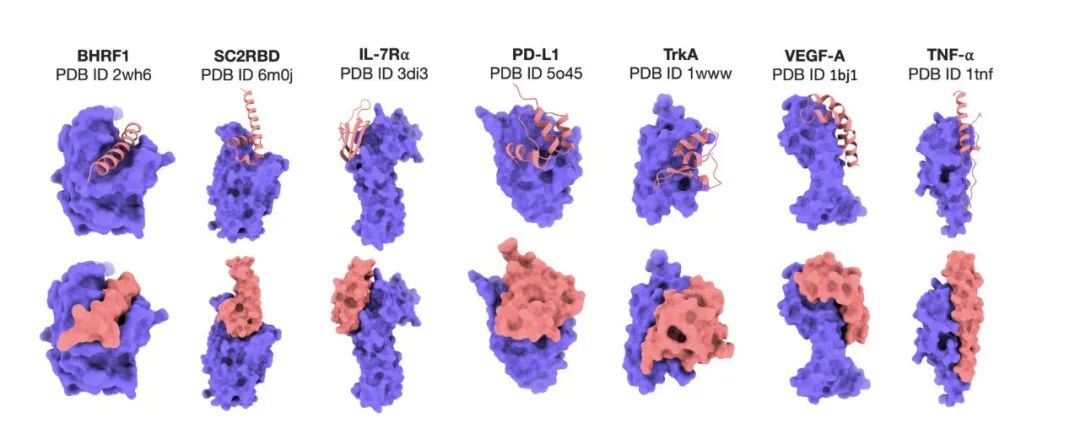
Um die Wirksamkeit des Prot42-Modells bei der Generierung von Proteinbindern genau zu bewerten, verglichen die Forscher das Modell mit AlphaProteo, einem fortschrittlichen Modell, das speziell für die Vorhersage von Proteinbindern entwickelt wurde. Die experimentellen Ergebnisse zeigten, dassDas Prot42-Modell generierte Bindemittel mit starken vorhergesagten Affinitäten zu mehreren therapeutisch relevanten Zielen.Insbesondere auf Zielmoleküle wie IL-7Rα, PD-L1, TrkA und VEGF-A,Das Prot42-Modell schnitt deutlich besser ab als das AlphaProteo-Modell.Diese Ergebnisse deuten darauf hin, dass das Prot42-Modell erhebliche Vorteile bei der Proteinbindererzeugung hat, wie in der folgenden Abbildung dargestellt.
DNA-sequenzspezifische Bindergenerierung
Auch im DNA-sequenzspezifischen Bindergenerierungsexperiment erzielte Prot42 bemerkenswerte Ergebnisse. Die experimentellen Ergebnisse zeigten, dassDurch die Kombination einer multimodalen Strategie aus Gen- und Proteineinbettung ist das Prot42-Modell in der Lage, Proteine zu erzeugen, die spezifisch an Ziel-DNA-Sequenzen binden und eine hohe Affinität aufweisen.Die vom DeepPBS-Modell ermittelte Bindungsspezifität war hoch. Diese Ergebnisse deuten darauf hin, dass das Prot42-Modell auch großes Potenzial für die Generierung DNA-sequenzspezifischer Binder besitzt und neue Werkzeuge für die Genregulation und Genomeditierung bietet.
Durchbrüche und Innovationen in der künstlichen Intelligenz im Proteindesign
Mit der tiefgreifenden Integration von Biotechnologie und künstlicher Intelligenz erfährt das bahnbrechende Forschungsfeld Proteindesign revolutionäre Veränderungen. Als zentrales Element lebender Prozesse war die Struktur- und Funktionsanalyse von Proteinen schon immer ein schwieriges Thema in der wissenschaftlichen Forschung. KI-Technologie beschleunigt die Lösung dieses komplexen Rätsels und eröffnet neue Wege für Szenarien wie die Entwicklung neuer Medikamente und die Transformation des Enzym-Engineerings.
In den letzten Jahren hat die KI-Technologie erneut Durchbrüche erzielt, und neue Technologien, die sich auf generative KI konzentrieren, bringen das Proteindesign in die „Genesis“-Phase.
Das Team von Professor Xu Dong an der University of Missouri schlug das strukturbewusste, proteinsprachenbewusste Modell (S-PLM) vor, das Proteinsequenz- und 3D-Strukturinformationen durch die Einführung von Multi-View-Kontrastlernen in einem einheitlichen latenten Raum ausrichtet.Wir verwenden Swin Transformer, um die von AlphaFold vorhergesagten Strukturinformationen zu verarbeiten und sie mit der ESM2-basierten Sequenzeinbettung zu verschmelzen, um ein strukturbewusstes PLM zu erstellen.Der Artikel „S-PLM: Strukturbewusstes Proteinsprachenmodell durch kontrastives Lernen zwischen Sequenz und Struktur“ wurde in Advanced Science veröffentlicht. S-PLM integriert Strukturinformationen geschickt in die Sequenzdarstellung, indem es Proteinsequenzen mit ihren dreidimensionalen Strukturen in einem einheitlichen latenten Raum ausrichtet. Darüber hinaus untersucht es effiziente Feinabstimmungsstrategien, die dem Modell eine hervorragende Leistung bei verschiedenen Proteinvorhersageaufgaben ermöglichen und einen wichtigen Fortschritt auf dem Gebiet der Proteinstruktur- und -funktionsvorhersage darstellen.
Papieradresse:
https://advanced.onlinelibrary.wiley.com/doi/10.1002/advs.202404212
Darüber hinaus schlugen das Forschungsteam der Tsinghua-Universität und andere das einheitliche Proteinsprachenmodell xTrimoPGLM vor. Dabei handelt es sich um ein einheitliches Vortrainingsframework und Basismodell, das auf 100 Milliarden Parameter erweitert werden kann und für verschiedene proteinbezogene Aufgaben, einschließlich Verständnis und Generierung (bzw. Design), konzipiert ist. Durch die Nutzung des allgemeinen Sprachmodells (GLM) als Grundlage seiner bidirektionalen Aufmerksamkeit und autoregressiven Ziele unterscheidet sich dieses Modell von früheren PLMs, die nur auf Encoder oder kausale Dekodierung basieren. Diese Studie untersuchte das einheitliche Verständnis und die Generierung vor dem Training von PLMs im ultragroßen Maßstab, eröffnete neue Möglichkeiten für das Proteinsequenzdesign und förderte die Weiterentwicklung eines breiteren Spektrums proteinbezogener Anwendungen. Sie wurde im Nature-Subjournal unter dem Titel „xTrimoPGLM: einheitlicher, auf 100 Milliarden Parameter vortrainierter Transformator zur Entschlüsselung der Proteinsprache“ veröffentlicht.
Papieradresse:
https://www.nature.com/articles/s41592-025-02636-z
Der Durchbruch von Prot42 ist nicht nur ein technischer Fortschritt, sondern spiegelt auch die zunehmende Reife des Modells „datengesteuertes + KI-Design“ im Bereich der Biowissenschaften wider. Zukünftig plant das Forschungsteam, die von Prot42 generierten Binder experimentell zu verifizieren und die computergestützte Auswertung durch tatsächliche Funktionstests zu ergänzen. Dieser Schritt wird den Nutzen des Modells in praktischen Anwendungen festigen und seine Vorhersagegenauigkeit verbessern und so die Lücke zwischen KI-gesteuerter Sequenzgenerierung und experimenteller Biotechnologie schließen.
Quellen:
1.https://arxiv.org/abs/2504.04453
2.https://mp.weixin.qq.com/s/SDUsXpAc8mONsQPkUx4cvA
3.https://mp.weixin.qq.com/s/x7_Wnws35Qzf3J0kBapBGQ
4.https://mp.weixin.qq.com/s/SDUsXpAc8mONsQPkUx4cvA








