Command Palette
Search for a command to run...
MIT Und Harvard Haben Gemeinsam PUPS Vorgeschlagen, Um Die Lokalisierung Einzelner Zellproteine Durch Die Integration Eines Proteinsprachenmodells Und Eines Bild-Inpainting-Modells Zu erreichen.
Die subzelluläre Lokalisierung eines Proteins bezieht sich auf die spezifische Position eines Proteins in der Zellstruktur.Dies ist für die Erfüllung biologischer Funktionen der Proteine von entscheidender Bedeutung. Um ein einfaches Beispiel zu geben: Stellen wir uns eine Zelle als ein riesiges Unternehmen vor, in dem Zellkern, Mitochondrien, Zellmembran usw. verschiedenen Abteilungen entsprechen, wie etwa dem Büro des Präsidenten, der Abteilung für Energieerzeugung und dem Gatekeeper. Nur wenn das entsprechende Protein in die richtige „Abteilung“ gelangt, kann es normal funktionieren, andernfalls verursacht es bestimmte Krankheiten wie Krebs und Alzheimer. Daher kann die präzise subzelluläre Lokalisierung von Proteinen als eine der Kernaufgaben der Biowissenschaften bezeichnet werden.
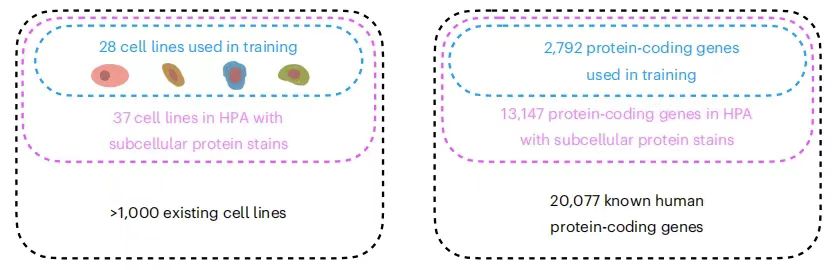
Obwohl die räumliche Lokalisierung von Tausenden von Proteinen in verschiedenen Zelllinien analysiert wurde, ist die Anzahl der bisher gemessenen Protein-Zelllinien-Kombinationen nur die Spitze des Eisbergs. Der derzeit größte verfügbare Datensatz zur subzellulären Lokalisierung ist beispielsweise:Der Human Protein Atlas (HPA) ermöglicht die subzelluläre Lokalisierung von Proteinen, die von 13.147 Genen kodiert werden (was 65% der bekannten menschlichen proteinkodierenden Gene entspricht).Der gesamte Datensatz umfasste jedoch 37 Zelllinien und jedes Protein wurde in höchstens drei davon gemessen. Gleichzeitig erschweren herkömmliche experimentelle Methoden die gleichzeitige Ermittlung der Anzahl aller Proteine in derselben Zelle. Dies erschwert die umfassende Analyse komplexer Proteinnetzwerke erheblich und erhöht die experimentelle Komplexität sowie das Fehlerrisiko.
Darüber hinaus ist die Proteinlokalisierung nicht statisch und ihre Variabilität tritt nicht nur zwischen Zelllinien, sondern auch zwischen einzelnen Zellen in der gleichen Zelllinie auf. Die in den vorhandenen Datenkarten erfassten Protein- und Zelllinienpaare spiegeln nur die Ergebnisse unter bestimmten Bedingungen wider. daher,Selbst vorhandene Ergebnisse lassen sich nur schwer direkt anwenden und es bedarf einer weiteren Erforschung der Proteinlokalisierung auf der Grundlage von Umweltveränderungen.
Um den Widerspruch zwischen den Beschränkungen der Methoden der subzellulären Proteinlokalisierung und der Komplexität biologischer Systeme aufzulösen, dürfte das maschinelle Lernen vielversprechend sein. Die heute entwickelten und erfolgreich eingesetzten Modelle, wie etwa proteinsequenzbasierte Modelle und zellbildbasierte Modelle, haben in einigen Aspekten gute Ergebnisse erzielt, weisen jedoch auch erhebliche Mängel auf: Erstere ignorieren die spezifischen Lokalisierungsunterschiede von Zelltypen und letztere verfügen nicht über die Generalisierungsfähigkeit, die für die Untersuchung unbekannter Proteine erforderlich wäre.
In Anbetracht dessenEin Forschungsteam des Massachusetts Institute of Technology und der Harvard University hat ein Vorhersagemodell für die subzelluläre Lokalisierung unbekannter Proteine vorgeschlagen, das auf der Kombination von Proteinsequenzen und Zellbildern basiert und den Namen Predictions of Unseen Proteins‘ Subcellular Localization (PUPS) trägt. PUPS kombiniert auf innovative Weise ein Proteinsprachenmodell und ein Bild-Inpainting-Modell, um die Proteinlokalisierung vorherzusagen. Dadurch ist es möglich, die Generalisierungsfähigkeiten unbekannter Proteinvorhersagen mit zelltypspezifischen Vorhersagen zu kombinieren, die die zelluläre Variabilität erfassen. Experimente haben gezeigt, dass dieses Framework die Lokalisierung von Proteinen in neuen Experimenten außerhalb des Trainingsdatensatzes genau vorhersagen kann, über hervorragende Generalisierungsfähigkeit und hohe Genauigkeit verfügt und ein herausragendes Anwendungspotenzial hat.
Die Forschungsergebnisse mit dem Titel „Prediction of protein subcellular localization in single cells“ wurden in Nature Methods veröffentlicht.
Forschungshighlights:
* Die vorgeschlagene Studie kombiniert auf innovative Weise Proteinsprachenmodelle und Bildwiedergabemodelle und verwendet Proteinsequenzen und Zellbilder, um die Proteinlokalisierung vorherzusagen und so die Mängel früherer Computermodelle auszugleichen.
* PUPS ist in der Lage, auf unbekannte Proteine und Zelllinien zu verallgemeinern, wodurch die Variabilität der Proteinlokalisierung zwischen Zelllinien und zwischen einzelnen Zellen innerhalb einer Zelllinie bewertet und biologische Prozesse identifiziert werden können, die mit Proteinen mit variabler Lokalisierung verbunden sind
* In neuen Experimenten außerhalb des Trainingsdatensatzes zeigte PUPS zudem seine hochpräzise Vorhersagefähigkeit mit herausragendem Anwendungspotenzial und medizinischem Wert

Papieradresse:
Datensätze: Aufbau vertrauenswürdiger Modelle mit möglichst umfassenden Daten
Der Trainingsdatensatz von PUPS stammt aus dem Human Protein Atlas (HPA).Das Forschungsteam hat die 16. Version der HPA-Daten in der 22. Version zusammengefasst, um möglichst viele Proteindaten zu sammeln und die Vollständigkeit der experimentellen Analyse sicherzustellen. Wie in der folgenden Abbildung dargestellt:
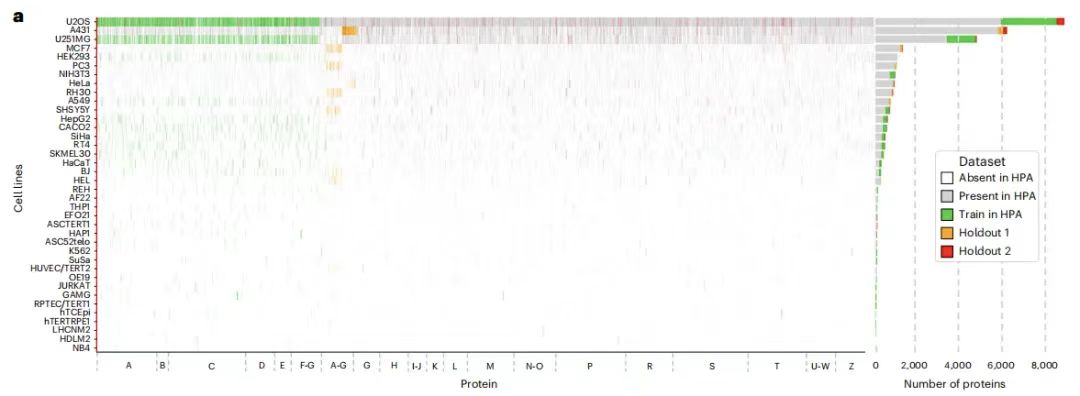
Konkret enthält der Trainingsdatensatz 340.553 Zellpopulationen mit insgesamt 8.086 Proteinvarianten, die 2.801 Genen in 37 Zelllinien in HPA entsprechen, deren Namen mit den Buchstaben AG beginnen. Darüber hinaus enthält der Trainingsdatensatz auch 10 zusätzliche Gene, darunter IHO1, IMPAD1, INKA1, ISPD, ITPRID1, KIAA1211L, KIAA1324, LRATD1, SCYL3 und TSPAN6.
Der Holdout-Datensatz ist in zwei Teile unterteilt:Ein Teil ist reservierter Datensatz 1,Es enthält 36.552 Zellen mit 9.472 Proteinvarianten, die 3.312 Genen entsprechen (davon 2.801 im Trainingssatz), deren Namen ebenfalls mit AG beginnen, aber aus verschiedenen Zelllinien stammen und keine Überschneidungen mit dem Trainingssatz aufweisen. In der Zwischenzeit wurde der zurückgehaltene Datensatz 1 weiter in zwei Teile aufgeteilt und als Auswertungssatz und Testsatz verwendet, der 11.050 bzw. 25.502 Zellen enthielt.Der beibehaltene Datensatz 2 enthält 24.007 Zellen, entsprechend 515 Genen.Sein Name beginnt mit allen Buchstaben des Alphabets und umfasst A-Z. Insgesamt gibt es 556 Proteinvarianten, die aus neuen Genfamilien stammen, die nicht im Trainingsset und im reservierten Datensatz1 vorkommen und zum Test der Generalisierungsfähigkeit des Modells verwendet werden können.
Es ist zu beachten, dass die Bilder der BJ-Zelllinie sowohl im Trainingssatz als auch im Holdout-Datensatz 1 beibehalten wurden.
Vor dem Experiment verarbeitete das Forschungsteam die Bilder in HPA vor, was im Wesentlichen die folgenden fünf Schritte umfasste:
* Schritt 1,Jedes Bild wurde viermal heruntergesampelt und die endgültige Auflösung auf 0,32 μm/Pixel reduziert, um den Rechenaufwand zu verringern und hochfrequentes Rauschen zu entfernen.
* Schritt 2,Gaußsche Unschärfe (σ=5) und Otsu-Schwellenwertbildung wurden kombiniert, um den ungefähren Bereich des Zellkerns vom komplexen Hintergrund zu trennen;
* Schritt 3, verwenden Sie die Funktion remove_small_holes, um Löcher mit einer Fläche kleiner als 300 Pixel zu entfernen, binarisieren Sie dann das Bild und entfernen Sie den Rauschbereich kleiner als 100 Pixel;
* Schritt 4,Der Schwerpunkt jedes Zellkerns wurde berechnet und ein 128 × 128 Pixel großer Bereich mit dem Schwerpunkt als ROI einer einzelnen Zelle ausgeschnitten;
* Schritt 5,Durch Intensitätsnormalisierung und Rauschfilterung wird eine standardisierte Datenverteilung erreicht und Interkanalstörungen werden reduziert.
Modellarchitektur: Kombination von Proteinsequenz und Bilddarstellung zur Vorhersage der subzellulären Proteinlokalisierung
Das PUPS-Modell besteht im Wesentlichen aus zwei Teilen:Eine wird verwendet, um die Sequenzdarstellung aus der Aminosäuresequenz des Proteins zu lernen; Die andere wird verwendet, um die Bilddarstellung aus der ikonischen Färbung der Zielzelle zu lernen.Die Proteinsequenzdarstellung und die Bilddarstellung werden dann kombiniert, um die subzelluläre Lokalisierung des Proteins in Zielzellen vorherzusagen. Ersteres ermöglicht die Verallgemeinerung des Modells auf Vorhersagen unbekannter Proteine, und Letzteres ermöglicht dem Modell, die Variabilität auf Einzelzellebene zu erfassen und zelltypspezifische Lokalisierungsvorhersagen zu treffen. Wie in der folgenden Abbildung dargestellt:
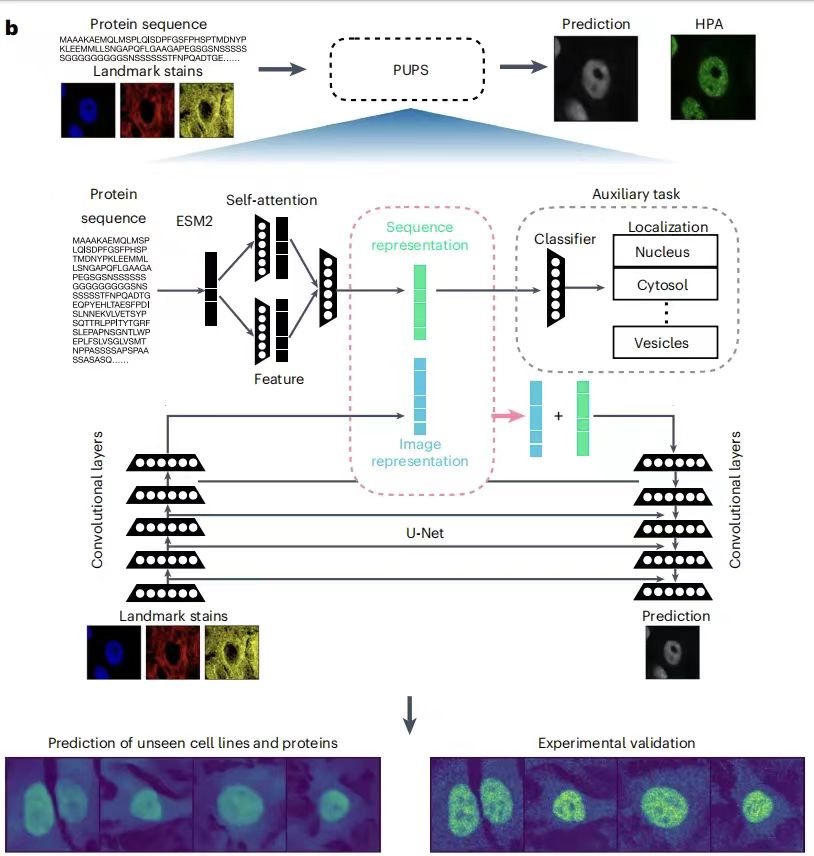
Einfach ausgedrückt:PUPS verwendet das vortrainierte Proteinsprachenmodell ESM-2 (Evolutionary Scale Modeling), um Protein-Sequenzmerkmale zu extrahieren, und verwendet ein Convolutional Neural Network, um die ikonischen Färbebildmerkmale von Zellen zu erlernen. Schließlich werden die beiden Informationsteile kombiniert, um die Lokalisierung von Proteinen in Zielzellen vorherzusagen.Es ist zu beachten, dass alle Teile des Modells gleichzeitig trainiert werden, was dazu beiträgt, den Klassifizierungsverlust der Voraufgabe und den Unterschied zwischen den vorhergesagten Proteinbildern und den experimentell gemessenen Proteinbildern in HPA zu reduzieren. Alle Parameter werden mit dem Adam-Optimierer mit einer Lernrate von 1e-4 optimiert.
Protein-Sprachmodell
PUPS lernt Sequenzdarstellungen mithilfe eines Sprachmodells, einer Selbstaufmerksamkeitsschicht und einer zusätzlichen Vortrainingsaufgabe und klassifiziert dann die Proteinlokalisierung basierend auf den erlernten Sequenzdarstellungen.
Konkret erhielt das Forschungsteam eine erste Darstellung einer bestimmten Proteinvariante, indem es die N-terminale 2.000 Aminosäuren umfassende Sequenz in das vorab trainierte ESM-2-Modell eingab und so für jeden Aminosäurerest einen 1.280-dimensionalen Vektor generierte, mit Null-Auffüllung für Varianten mit weniger als 2.000 Resten. Dieser Grenzwert für die Sequenzlänge soll verzerrte Vorhersagen für die wenigen Proteine mit Sequenzlängen von bis zu Zehntausenden von Resten vermeiden. Wie in der folgenden Abbildung dargestellt:
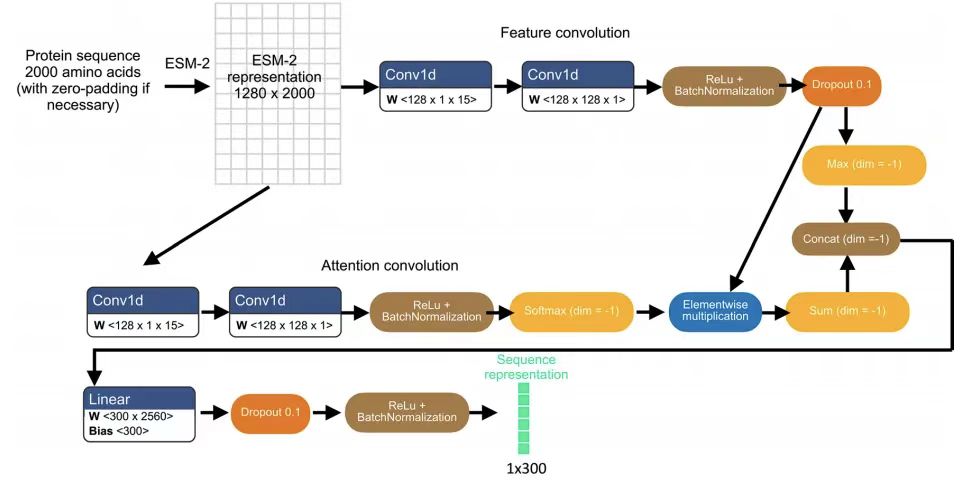
Um die ESM-2-Charakterisierung für die Vorhersage der Proteinlokalisierung anzupassen,Anschließend übernahm das Team eine leichte Aufmerksamkeitsschicht aus trennbaren Faltungen.Angewandt auf die ESM-2-Darstellung erhält man schließlich eine 300-dimensionale Sequenzdarstellung. Diese Proteinsequenzdarstellung wird sowohl für die zusätzliche Voraufgabe der Vorhersage von Lokalisierungsmarkierungen als auch für die Proteinbildvorhersage in Kombination mit der Bilddarstellung verwendet. Die Voraufgabe gibt die Protein-Sequenzdarstellung in eine vollständig verbundene neuronale Netzwerkschicht ein, um einen 29-dimensionalen Vektor einzugeben, der die Wahrscheinlichkeitsverteilung unter 29 subzellulären Kompartimentlokalisierungsbezeichnungen darstellt, und vergleicht dann die Voraufgabe-Ausgabe mit den HPA-annotierten Proteinkompartimenten unter Verwendung eines binären Kreuzentropieverlusts mit Sigmoid-Aktivierung.
Bild-Rendering-Modell
Der Bildeingang jeder Zelle enthält drei ikonische Färbungsbildkanäle: Zellkern-, Mikrotubuli- und endoplasmatische Retikulum-Färbung.Seine Abmessungen betragen 3 x 128 x 128 und sind auf den Kernschwerpunkt zentriert.
Die Bildkodierung wird durch 5 trennbare Faltungsschichten erreicht.Endmaße 16 x 16 x 512 . Auf jede Faltungsschicht folgen LeakyRelu-Aktivierung, Batch-Normalisierung und 2D-Max-Pooling-Schichten. Die Proteinsequenzdarstellung wird mit allen räumlichen Dimensionen der Zellbilddarstellung verknüpft und dann in einen U-Net-Bilddecoder eingespeist, der für jeden Eingangskanal unterschiedliche Gewichte lernt. Darüber hinaus ermöglicht der Mechanismus zur Gewichtung räumlicher Dimensionen im Modell, jede räumliche Dimension der Bilddarstellung mit unterschiedlichen Gewichtungen mit der Sequenzdarstellung zu kombinieren.
Der Decoder besteht aus 5 trennbaren Faltungsschichten.Generiert eine 1 x 128 x 128 Bildausgabe, die die Proteinbildvorhersage für die entsprechende Zelle ist. Anschließend werden ähnliche Sprungverbindungen wie im Bildsegmentierungs-U-Net zwischen der Codierungsschicht, die Bilddarstellungen der Landmark-Färbung generiert, und der Decodierungsschicht, die Proteinbildvorhersagen in derselben Tiefe generiert, hinzugefügt. In der Studie wurde eine mittlere quadratische Fehlerverlustfunktion verwendet, um das Modell zu trainieren und den Unterschied zwischen den vorhergesagten Proteinbildern und den experimentell gemessenen Proteinbildern zu minimieren.
Experimentelle Ergebnisse: Präzise subzelluläre Lokalisierung von Proteinen auf Einzelzellebene
Um die Machbarkeit und Wirksamkeit des Modells zu überprüfen, schlug das Forschungsteam eine Reihe von Experimenten zur Überprüfung vor. PUPS zeigte bei mehreren Aufgaben eine gute Leistung und hob seine Vorteile bei der Multimodellfusion hervor.
Vorhersage der Variabilität der Proteinlokalisierung zwischen Zelllinien
Um die Leistung von PUPS bei der Quantifizierung der Proteinlokalisierungsvariabilität zwischen Zelllinien zu bewerten,Das Forschungsteam berechnete das Protein-Kern-Verhältnis, um die Lokalisierungsvariabilität zu quantifizieren, und stellte fest, dass die vorhergesagten Werte stark mit den tatsächlichen Daten korrelierten.Der Pearson-Korrelationskoeffizient für Holdout 1 beträgt 0,794 und der Pearson-Korrelationskoeffizient für Holdout 2 beträgt 0,878. Wie in der folgenden Abbildung dargestellt:
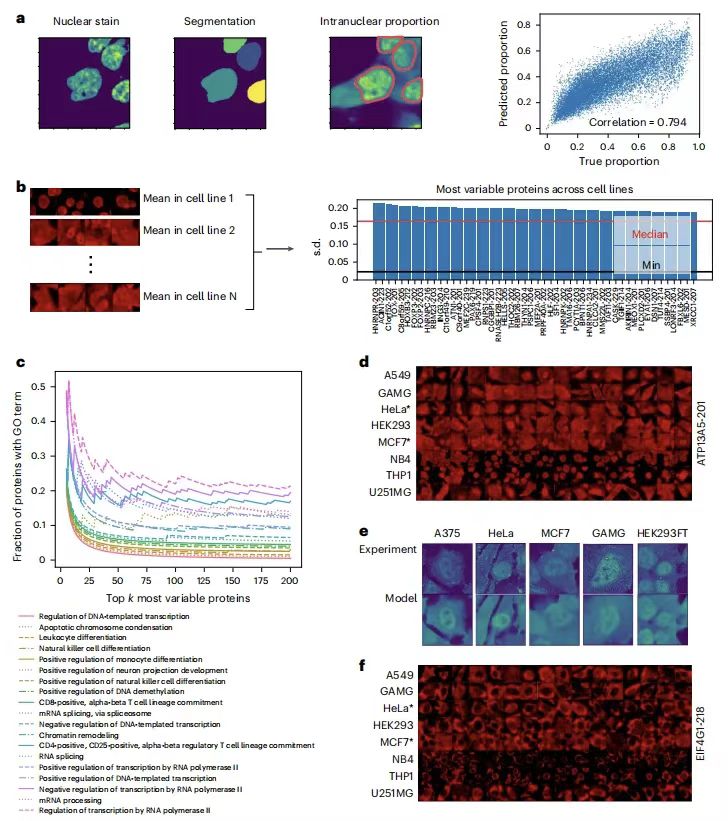
Nachfolgende weitere Analysen zeigten, dass die Proteine mit den größten Lokalisierungsänderungen zwischen Zelllinien mit biologischen Prozessen wie Transkription, Zelldifferenzierung und Chromatinregulation in Zusammenhang standen. Die experimentelle Validierung von ATP13A5 bestätigte die Genauigkeit der Vorhersagen des Modells. Auch,Das Modell erfasst Unterschiede in der Zellmorphologie durch Signaturfärbung und kann auf die Zelllinienspezifität der Proteinlokalisierung ohne Zelllinienmarkierungen schließen, und bietet eine neue Methode zur Untersuchung der zellspezifischen Regulierung der Proteinfunktion.
Unterschiede in der Proteinlokalisierung zwischen einzelnen Zellen vorhersagen
Um die Fähigkeit von PUPS zu bewerten, die Variabilität der Proteinlokalisierung zwischen einzelnen Zellen in der gleichen Zelllinie vorherzusagen, berechnete das Forschungsteam die Varianz des nukleären Verhältnisses von Proteinen in allen einzelnen Zellen in jeder Zelllinie.Die Ergebnisse zeigten, dass die Rangfolge der Vorhersage der Einzelzellvariabilität für jedes Protein-Zelllinienpaar in hohem Maße mit den tatsächlichen Daten übereinstimmte.Beispielsweise überschritt die Überlappungsrate der ersten 500 hochvarianten Paare in Holdout 2 60%, und die vorhergesagte intranukleäre Verhältnisverteilung stimmte mit den tatsächlichen Ergebnissen überein, wodurch der Einfluss von Vorhersagefehlern eliminiert wurde.
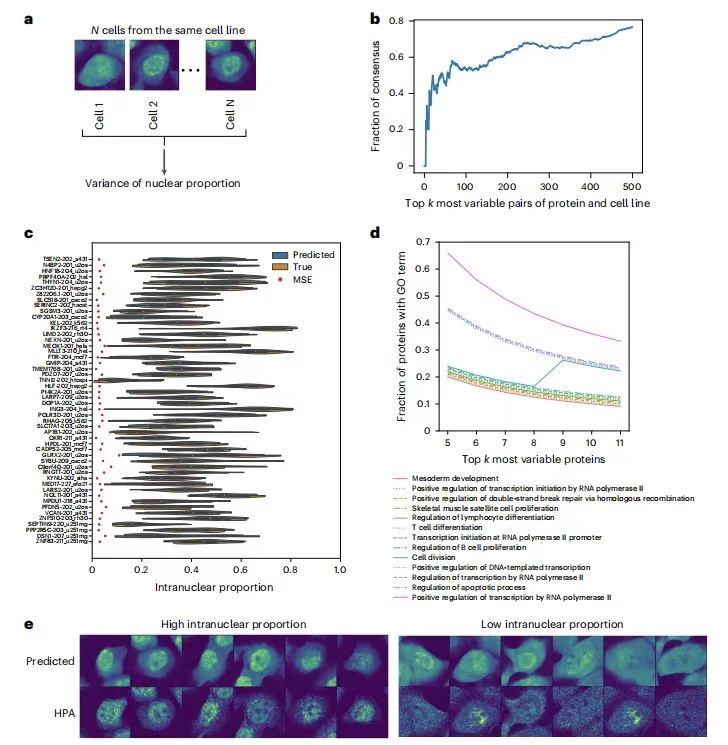
Darüber hinaus zeigte die Genontologie-Analyse (GO), dass hochvariable Proteine mit Prozessen wie Zellteilung, Transkription, Reparatur von Doppelstrangbrüchen und Apoptose in Zusammenhang stehen. Auch,Das Modell erfasst morphologische Merkmale durch Zellmarkierungs-Färbungsbilder und weist darauf hin, dass die Variabilität einzelner Zellen nicht nur zufällig ist, sondern auch mit morphologischen Zellmerkmalen zusammenhängt.Bietet eine neue Perspektive zur Erklärung des Mechanismus der Einzelzellheterogenität.
Validierung von PUPS in neuen Experimenten außerhalb der Trainingsdaten
Um die Ubiquitinierungsfähigkeit von PUPS zur Vorhersage der Proteinlokalisierung in neuen experimentellen Umgebungen zu überprüfen, wählte das Forschungsteam 9 Proteine zur Überprüfung in 5 Zelllinien aus. Wie in der folgenden Abbildung dargestellt:
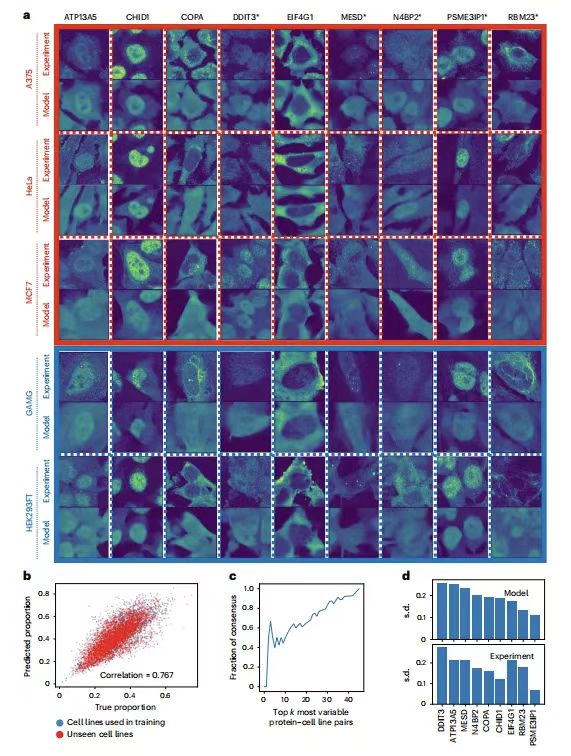
ATP13A5, CHID1, COPA, MESD und RBM23 sind die Proteine mit der größten Variation zwischen den Zelllinien und sie alle haben unterschiedliche GO-Begriffe; DDIT3 und N4BP2 sind die Proteine mit der größten Variation in einzelnen Zellen innerhalb einer Zelllinie; EIF4G1 und PSME3IP1 sind die Proteine mit der geringsten Variation zwischen den Zelllinien. Ersteres dürfte sich hauptsächlich außerhalb des Zellkerns befinden, Letzteres dürfte sich hauptsächlich innerhalb des Zellkerns befinden. Von den fünf Zelllinien sind außer A375 die anderen HeLa, MCF7, GAMG und HEK293FT in der HPA enthalten.
Die Ergebnisse zeigen, dassDie von PUPS vorhergesagten Proteinbilder ähneln optisch den experimentell gemessenen.Das anhand des vorhergesagten Proteinbildes berechnete Kernproteinverhältnis jeder einzelnen Zelle korreliert eng mit dem aus dem experimentell gemessenen Bild berechneten Verhältnis, mit einem Pearson-Korrelationskoeffizienten von 0,767. Dies zeigt, dassMit PUPS kann die Lokalisierung von Proteinen quantitativ vorhergesagt werden, die zuvor nicht experimentell gemessen oder in Trainingsatlanten verwendet wurden.
PUPS lernt aussagekräftige Protein- und Zelldarstellungen
Experimente zeigen, dass die Fähigkeit von PUPS, die Proteinlokalisierung in unbekannten Proteinen und Zelllinien vorherzusagen, auf dem Erlernen aussagekräftiger Darstellungen von Proteinsequenzen und Zelllandmarkenbildern beruht.
Das Forschungsteam kartierte Proteinsequenzdarstellungen von 40.622 Proteinformen, die 12.614 Genen entsprechen, und Proteine mit ähnlicher Lokalisierung neigten dazu, ähnliche Sequenzdarstellungen aufzuweisen. Um weiter zu demonstrieren, dass das Modell aussagekräftige Protein-Sequenzmuster identifizieren und die Lokalisierung vorhersagen kann, berechnete das Forschungsteam mithilfe der Positional-Shapley-Methode die Bedeutung jedes Aminosäurerests in einem bestimmten Protein für die Vorhersage der Markierungen jedes Zellkompartiments. So konnte beispielsweise die vorhergesagte Variabilität der nukleären Lokalisierung von N4BP2 erfolgreich erklärt werden, was mit Berichten übereinstimmt, denen zufolge die CUE-Domäne die subzelluläre Lokalisierung durch Ubiquitinbindung verändern könnte.
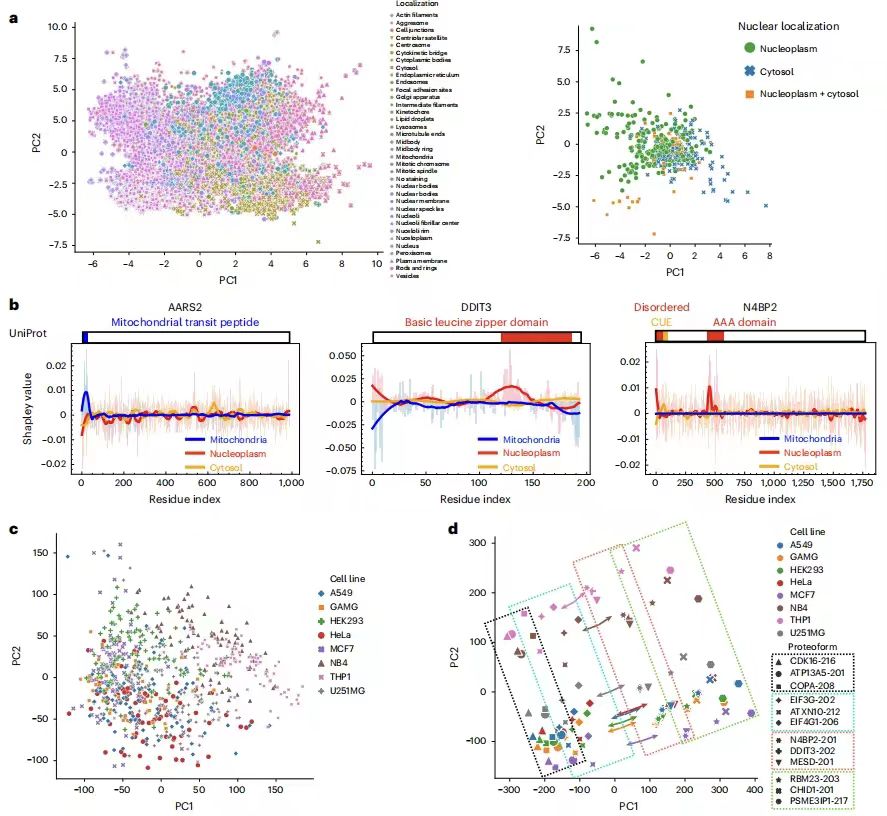
Neben der Identifizierung bedeutsamer Proteinsequenzmotive,Das Forschungsteam zeigte außerdem, dass PUPS aus der Zellsignaturfärbung aussagekräftige Darstellungen einzelner Zellen lernt.Es visualisiert die durch Landmark-Färbung gewonnenen Bilddarstellungen einzelner Zellen und stellt fest, dass einzelne Zellen derselben Zelllinie ähnliche Bilddarstellungen aufweisen, auch wenn die Zelllinienbezeichnung nicht in das Modell eingegeben wird. Durch die gemeinsame Darstellung von Proteinen und Zelllandmarkenbildern bleibt die Trennung zwischen Zelllinien und Proteinen erhalten, während die Reihenfolge der verschiedenen Proteine innerhalb jeder Zelllinie über alle Zelllinien hinweg ähnlich ist. Bei gegebenem Schwerpunkt jeder Zelllinie im gemeinsamen Darstellungsraum sind die Vektoren vom Schwerpunkt zu einem bestimmten Protein über alle Zelllinien hinweg größtenteils parallel, d. h. bei gegebener Sequenzdarstellung erfordert die Vorhersage eines Bildes für ein bestimmtes Protein, dass man sich im Darstellungsraum unabhängig von der Zelllinie in dieselbe Richtung bewegt.Dies erklärt die Fähigkeit von PUPS, durch das Erlernen aussagekräftiger Darstellungen von Protein- und Zellbildern auf unbekannte Proteine und Zelllinien zu verallgemeinern.
Auch,PUPS kann auch die Auswirkungen krankheitsverursachender Mutationen auf die Proteinlokalisierung vorhersagen.So zeigten etwa Mutationsstudien an den nukleär kodierten mitochondrialen Proteinen SDHD und ETHE1, dass SDHD-Mutationen zu einer Erhöhung ihres Kernlokalisierungsverhältnisses führen, was mit dem Mechanismus der nukleären Genominstabilität bei der Erkrankung übereinstimmt. ETHE1-Mutationen zeigen eine Erhöhung des zytoplasmatischen Lokalisierungsverhältnisses, was mit bekannten Anomalien des nuklear-zytoplasmatischen Shuttles verbunden ist. Diese Ergebnisse legen nahe, dass PUPS durch die Analyse der Auswirkungen von Sequenzvariationen auf die Lokalisierung neue Hinweise für die Untersuchung von Krankheitsmechanismen liefern kann.
Neue Lösung zur Vorhersage der subzellulären Proteinlokalisierung
Wie oben erwähnt, ist die Vorhersage der subzellulären Lokalisierung von Proteinen sowohl in der Bioinformatik als auch in der biologischen Forschung von großer Bedeutung. PUPS bietet eine Möglichkeit zur Integration multimodaler Informationen, was der Forschung auf diesem Gebiet einen wichtigen Impuls verleiht. Gleichzeitig hat die Forschung auf diesem Gebiet nach Jahrzehnten der Entwicklung eine große Vielfalt an Ergebnissen hervorgebracht.
Ein Team des University College Dublin in Irland veröffentlichte eine Studie im Computational and Structural Biotechology Journal.Es werden verschiedene rechnergestützte Methoden zur Vorhersage der subzellulären Lokalisierung von Proteinen vorgestellt, darunter sequenzbasierte, annotationsbasierte, hybride und auf Metavorhersagen basierende Methoden. Der Artikel klassifiziert und stellt außerdem Tools zur Vorhersage der subzellulären Lokalisierung nach Eukaryoten, Prokaryoten, Viren und mehreren Kategorien vor.Zu den eukaryotischen Vorhersagetools gehören mLASSO-Hum, DeepPSL usw., und zu den prokaryotischen Vorhersagetools gehören PRED-LIPO usw. Durch die Entwicklung einer Klassifizierungskarte für maschinelles Lernen und Deep Learning, die 7 Hauptbereiche und 28 Unterkategorien abdeckt, bietet diese Studie eine Taxonomie von Vorhersagetools mit einer und mehreren Kategorien, wodurch Benutzer Methoden und Vorhersagetools leichter finden können. Der Artikel wurde unter dem Titel „Protein subcellular localization prediction tools“ veröffentlicht.
* Papieradresse:
https://www.sciencedirect.com/science/article/pii/S2001037024001156
Am 12. April veröffentlichten die Forschungsgruppe von Yang Li am Institute of Biomedical Sciences der Universität Fudan und die Forschungsgruppe von Dong Nanqing am Shanghai Artificial Intelligence Laboratory gemeinsam ein Online-Forschungspapier mit dem Titel „Deep Generative Model for Protein Subcellular Localization“ in der Zeitschrift Briefings in Bioinformatics.Die Studie entwickelte außerdem ein generatives Deep-Learning-Modell deepGPS mit multimodalen Verarbeitungsfunktionen basierend auf dem ESM2-Proteinsprachenmodell und dem U-Net-Framework.
Berichten zufolge kann deepGPS Proteinsequenzen und Zellkernbilder als Eingabe empfangen und Textbeschriftungen und Verteilungsbilder der Proteinlokalisierung generieren. Es handelt sich um ein neues multimodales „Text-zu-Bild“-Modell, das die Vorhersage der subzellulären Lokalisierung von Proteinen unterstützt.
* Papieradresse:
https://doi.org/10.1093/bib/bbaf152
Mit der zunehmenden Integration von künstlicher Intelligenz und biologischer Forschung entstehen ständig neue innovative Experimente, die die Nachteile traditioneller Methoden schrittweise überwinden und „das Beste aus beiden Welten“ oder sogar eine „perfekte“ Leistung erzielen, wodurch die rasante Entwicklung der Bioinformatik gefördert wird.








