Command Palette
Search for a command to run...
Zum Ersten Mal Schlug Ein Team Der Columbia University PXRDnet Zur End-to-End-Analyse Von Nanokristallen Vor Und Analysierte Erfolgreich 200 Komplexe Simulierte Nanokristalle.

Die Entdeckung und Anwendung der Röntgenbeugung (XRD) ist ein wichtiger Meilenstein in der Entwicklung der Kristallographie, da diese Technologie es ermöglicht, ein tiefes Verständnis der Mikrostruktur von Kristallen zu erlangen, was wiederum den Fortschritt der Materialwissenschaften und der menschlichen Zivilisation als Ganzes vorantreibt. Bei der Anwendung von pulverförmigen Nanokristallen aus winzigen Partikeln werden mit herkömmlichen Methoden jedoch nicht die gewünschten Ergebnisse erzielt.
Aufgrund der begrenzten Größe von Nanokristallen (normalerweise weniger als 1000 Å),Der Bragg-Peak in seinem Röntgenbeugungsmuster zeigt eine deutliche Verbreiterung.Dies führt zu einer erheblichen Verschlechterung der Strukturinformationen, was die genaue Auflösung der Kristallstruktur zu einer enormen Herausforderung macht. Darüber hinaus erschwert die Schwierigkeit, in der Praxis reine Einkristallproben zu erhalten, die Strukturanalyse zusätzlich. Die Analyse der Nanokristallstruktur ist zudem zu einem „jahrhundertealten Problem“ geworden, das die Materialwissenschaftsgemeinschaft seit hundert Jahren plagt.
Um dieses Problem zu lösen, schlugen Forscher der Columbia University und der Stanford University eine generative Methode zur Strukturanalyse mit künstlicher Intelligenz namens PXRDnet vor, die auf einem Diffusionsmodell basiert.Das Modell verwendet 45.229 bekannte Kristallstrukturen als Trainingsdaten und führt statistisches Vorwissen ein.Obwohl lediglich die chemische Formel und das informationsarme, verbreiterte Pulverbeugungsmuster mit begrenzter Größe als Bedingungen dienten, konnte PXRDnet 200 simulierte Nanokristalle unterschiedlicher Symmetrie und Komplexität erfolgreich auflösen.Es sind Strukturen aus allen sieben Kristallsystemen bis hinunter zu einer Größe von 10 Å enthalten.Experimentelle Ergebnisse zeigen, dass das Modell 4 von 5 Strukturkandidaten erfolgreich und nachweisbar identifizieren kann, mit einem durchschnittlichen Fehler von nur 7% nach der Messung mit dem Rietveld-Verfeinerungsfaktor r.
Die entsprechende Forschung wurde in Nature Materials unter dem Titel „Ab initio structure solutions from nanocrystalline powder diffraction data via diffusion models“ veröffentlicht.
Forschungshighlights:
* Diese Errungenschaft hat das langjährige Problem der Nanokristallstrukturanalyse in der Materialwissenschaftsgemeinschaft gelöst und ein effizientes Analysetool mit künstlicher Intelligenz bereitgestellt, das voraussichtlich innovative Anwendungen in der Nanotechnologie, Biomedizin, Energiespeicherung, elektronischen Geräten und anderen Bereichen fördern wird.
* Diese Methode durchbricht die Anwendungsgrenzen traditioneller Methoden erheblich und liefert in vielen Fällen Kandidatenlösungen, die der realen Struktur nahe kommen
* Die Studie schlug den MP-20-PXRD-Benchmark-Datensatz vor (einschließlich stabiler Materialien mit weniger als 20 Atomen im Materials Project und ihrer simulierten Beugungsdaten) und machte den Code und den Datensatz öffentlich, wodurch ein einheitlicher Standard für nachfolgende Forschungen geschaffen wurde

Papieradresse:
https://go.hyper.ai/r1K6b
Online-Materialdatenbank des Materials Project:
https://go.hyper.ai/2gCe9
Datensatz: Vorgeschlagener MP-20-PXRD-Benchmark-Datensatz
Um ein effektives Modell zu erhalten, stellten die Forscher einen Benchmark-Datensatz namens MP-20-PXRD für das End-to-End-Training von PXRDnet bereit.
Konkret nutzten die Forscher den MP-20-Datensatz des Materials Project.Der Datensatz besteht aus Materialien, die aus der Materials Project-Datenbank entnommen wurden und maximal 20 Atome in der Elementarzelle aufweisen.Anschließend verwendeten die Forscher das Pymatgen-Paket, um die Pulverbeugungsmuster aller Strukturen in MP-20 zu simulieren.
Online-Materialdatenbank des Materials Project:
https://go.hyper.ai/2gCe9
Die Simulationen verwendeten Cu Kα-Strahlung mit einem Q-Bereich von 0-8,1568 Å⁻¹.
Der MP-20-PXRD-Datensatz enthält 45.229 Materialien.Die Verhältnisse von 90%, 7,5% und 2,5% werden zum Trainieren, Verifizieren und Testen verwendet. Es ist erwähnenswert, dass der MP-20-PXRD-Datensatz als Open Source zur Verfügung gestellt wurde und die Forscher hoffen, ihn nutzen zu können, um „Nachzügler“ zu inspirieren, neue Lösungen für die Nanokristallstrukturanalyse weiter zu erforschen.
Modellarchitektur: Basierend auf CDVAE, Einführung des PXRD-Regressors
Das PXRDnet-Modell basiert auf der CDVAE-Architektur.Es besteht im Wesentlichen aus drei Hauptzweigen, nämlich dem Zweig der atomaren Rauschunterdrückung, dem Zweig des Variational Autoencoder (VAE) und dem PXRD-Regressor.Sie sind über einen gemeinsamen latenten Gauß-Code verbunden. Dieser Ansatz ermöglicht es PXRDnet, anhand eines PXRD-Musters und einer chemischen Formel qualifizierte Materialstrukturkandidaten präzise zu generieren und so neue Einblicke in die Strukturanalyse von Nanomaterialien zu gewinnen.
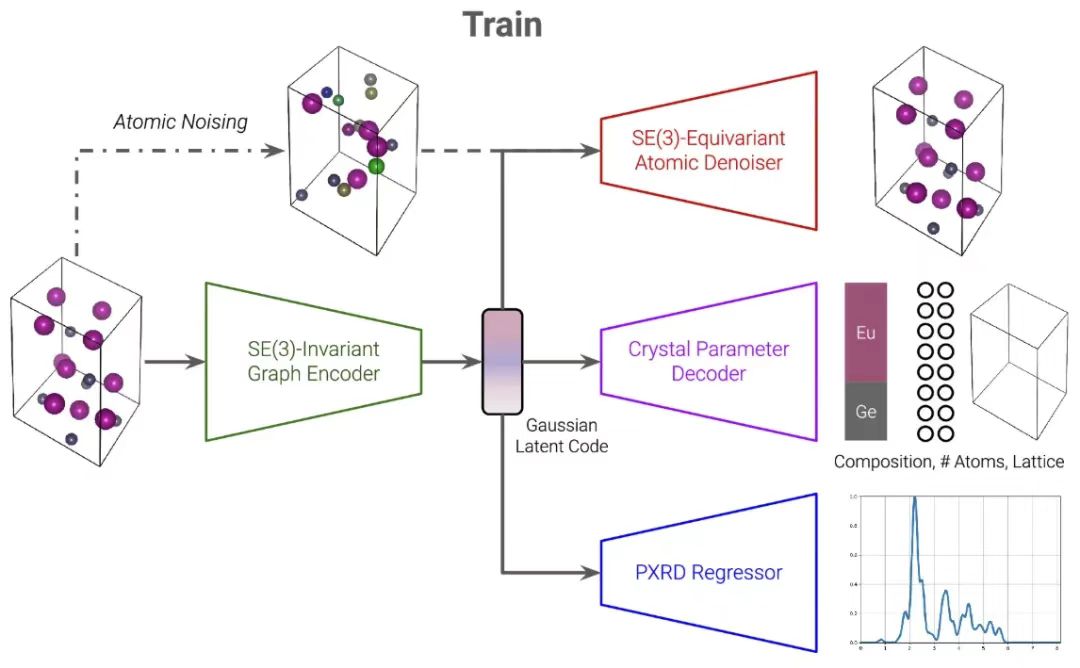
Skelettentwicklung basierend auf CDVAE
Bei der Einführung von PXRDnet müssen wir das CDVAE-Modell erwähnen, das die Grundlage für die Erstellung des ersteren bildet.CDVAE ist ein Modell zur Generierung von Materialstrukturen.Es ist von Variational Autoencodern und Denoising Diffusion Networks inspiriert und ist ein generatives Modell, das lernt, Daten von Rauschen zu befreien.
Um die Zerlegung der VAE- und Diffusionskomponenten zu verstehen, erkannten die Forscher, dass die Elementarzelle eines Materials durch vier Komponenten dargestellt werden kann: chemische Zusammensetzung, Anzahl der Atome, Gitterparameter und Atomkoordinaten.
Der erste Zweig von CDVAE verwendet VAE, um die ersten drei Komponenten zu verarbeiten.Der Encoder ist DimeNet, ein SE(3)-invariantes Graph Neural Network, das die Graphdarstellung des Materials auf eine latente Darstellung z abbildet. Die Graphdarstellung wird in einen gerichteten Multigraphen geändert, um die inhärente Periodizität des Materials widerzuspiegeln. Anschließend regularisierten die Forscher die latente Darstellung z mithilfe des Kullback-Leibler-Divergenzverlusts in eine multivariate Gauß-Verteilung und dekodierten anschließend die chemische Zusammensetzung, die Ordnungszahl und die Gitterparameter von z.
Jede Vorhersage wird von einem separaten kristallparametrisierten Multilayer-Perceptron (MLP) generiert, das den latenten Code z empfängt.z wird als Materialdarstellung in allen anderen Zweigen des nachfolgenden Modells verwendet.
Der zweite Zweig von CDVAE nutzt die Rauschunterdrückungsdiffusion, um Komponenten durch ein rauschkonditioniertes Score-Netzwerk zu verarbeiten.Dabei wird davon ausgegangen, dass die Anzahl der Atomkomponenten und die Gitterparameter fest sind. Der Vorwärtsprozess verwendet multivariates Gaußsches Rauschen, um die Atomkoordinaten und Atomarten zu stören. Der umgekehrte Prozess wird mit GemNet, einem SE(3)-äquivarianten Graph-Neuralnetzwerk, parametrisiert. Dieser Prozess ist an den oben beschriebenen latenten Code z gebunden, der die Grundlage für seinen normalen Ablauf bildet.
Es ist erwähnenswert, dassDer umgekehrte Prozess besteht im Wesentlichen darin, vorherzusagen, wie die gestörten Atomkoordinaten und -spezies über die Langevin-Dynamik entrauscht werden können.Bringen Sie sie dazu, an ihren wahren Standort zurückzukehren und ihre wahre Art wiederherzustellen. Ebenso ist die Ausgabegraphdarstellung ein gerichteter Multigraph, der mit der Periodizität des Materials kompatibel ist.
In der Generierungsphase wählt CDVAE zunächst einen latenten Code z ≈ N (0, I) aus einer multivariaten Gauß-Verteilung aus.Zur Dekodierung wird das mehrschichtige Kristallparameter-Perzeptron verwendet, um die chemische Zusammensetzung der Komponente, die Ordnungszahl und die Gitterparameter zu ermitteln, die zur Initialisierung einer Einheitszelle verwendet werden können, wobei die Atompositionen ebenfalls zufällig aus N (0, I) ausgewählt werden. Die Atompositionen und -typen werden dann durch den äquivarianten Bildentrauschungsprozess der Langevin-Dynamik SE (3) optimiert. Während des gesamten Entrauschungsprozesses bleiben die Gitterparameter und die Ordnungszahl unverändert und schließlich wird das resultierende Material erhalten.
Speziell entwickelter PXRD-Regressor
Darüber hinaus wurde in dieser Studie das Pulver-Röntgenbeugungsmuster (PXRD) als die gewünschte vorherzusagende Eigenschaft festgelegt, sodass die Forscher einen PXRD-Regressor Fψ entwickelten, der die potenzielle Materialdarstellung z∈R transformiert256 Abgebildet auf einen Vektor y∈R512, d. h. die geschätzte Q-Raum-Charakterisierung des PXRD-Musters des Materials.
Der PXRD-Regressor wird durch eine von DenseNet inspirierte Architektur parametrisiert.Diese Architektur erweitert das traditionelle Convolutional Neural Network.Der Regressor basiert auf dem Design von CrystalNet mit einer dicht verbundenen Architektur mit eindimensionaler Eingabe und Ausgabe. Insbesondere gilt für eine gegebene Tiefe im Netzwerk:DenseNet aggregiert vorherige Zwischendatendarstellungen als Eingabe für die nächste Faltungsschicht.Wie in der Abbildung unten gezeigt.
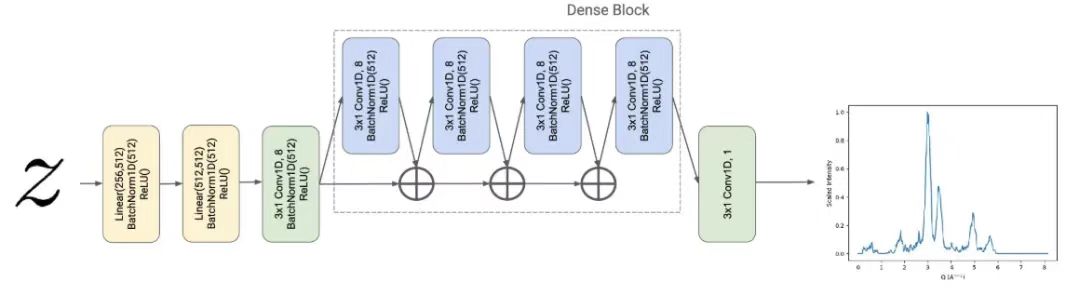
Untersuchungen haben gezeigt, dass DenseNet das Problem des verschwindenden Gradienten reduziert und bei Standard-Computer-Vision-Benchmarks hervorragende Ergebnisse erzielt.
Experimentelle Ergebnisse: Potenzial für die Anwendung in der Praxis
Normalerweise werden Nanostrukturen als Kristalle mit einer Größe von weniger als 1000 Å definiert. Um jedoch die Wirksamkeit der vorgeschlagenen Methode zu testen, reduzierten die Forscher die Größe der Kristalle um zwei Größenordnungen, indem sie die PXRD-Methode mit Kristallgrößen von 10 Å und 100 Å mithilfe einer mathematischen Filtermethode auf Basis der Fourieranalyse simulierten. Wie erwartet,Im 10-Å-Fall ist die Peakverbreiterung größer als im 100-Å-Fall, was darauf hinweist, dass die Informationsverschlechterung stärker bestätigt ist.Wie in der Abbildung unten gezeigt.
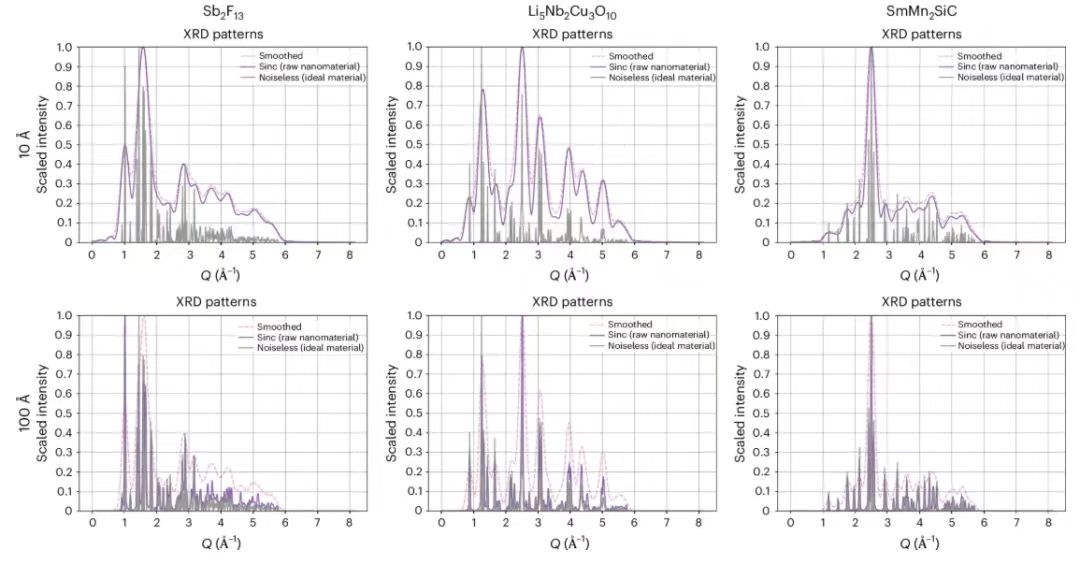
Dieses Bild zeigt, wie Forscher die Auswirkungen der Schrumpfung im Nanobereich auf PXRD-Spitzen mithilfe der Sinc²-Filterung simulierten. Dabei stellt die graue Linie den idealen Modus dar und die violette Linie den PXRD-Peak, der nach der Behandlung verbreitert ist.Um die Modellleistung zu verbessern, haben die Forscher nach dem Sinc-Filter zusätzlich einen Gauß-Filter angewendet.Dadurch wird zwar die Verbreiterung der Beugungsspitzen verstärkt, die durch die Filterung verursachten scharfen Wellen können jedoch wirksam eliminiert werden. Die horizontale Achse stellt die Größe des Streuvektors in Å⁻¹ dar und die vertikale Achse stellt die skalierte Beugungsintensität dar, wobei 1 den maximalen Intensitätswert darstellt.
Als nächstes präsentierten die Forscher die PXRDnet-Strukturvorhersage, wie unten gezeigt. Die Spalte ganz links zeigt die tatsächliche Kristallstruktur, und die anderen Spalten zeigen die rekonstruierten Kristallstrukturen von Nanokristallen mit Durchmessern von 10 Å und 100 Å, die von PXRDnet im PXRD-Muster nach Rietveld-Verfeinerung simuliert wurden.
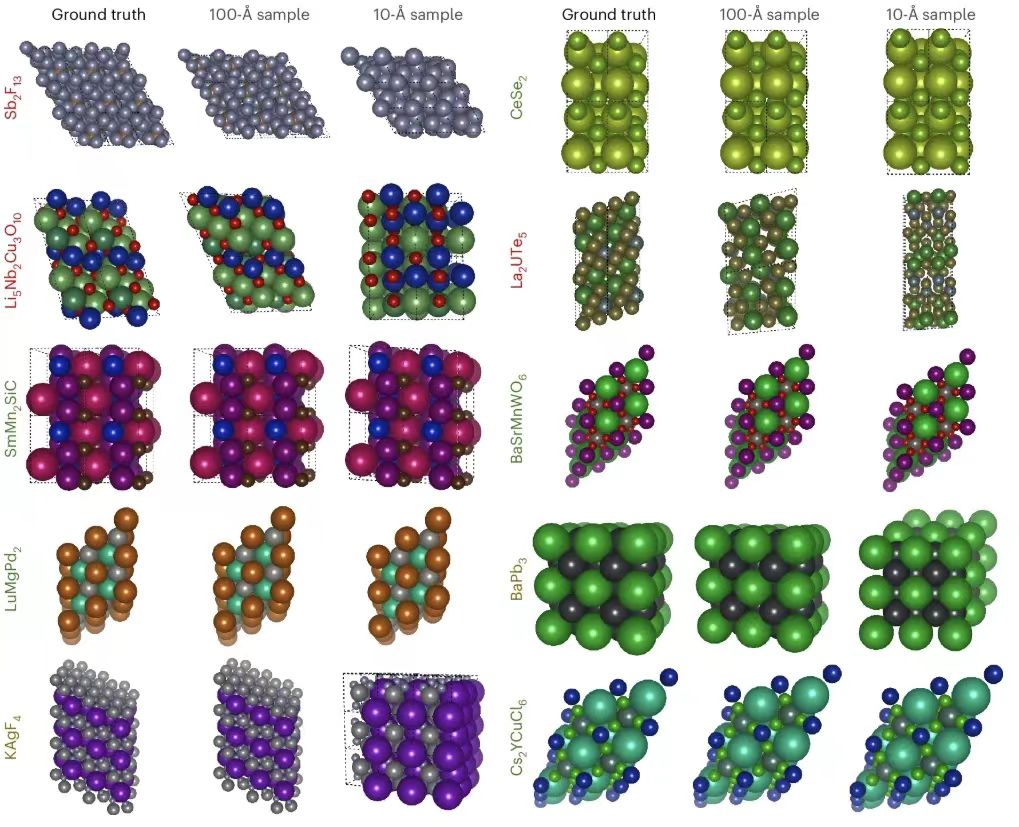
Die Ergebnisse zeigen, dass PXRDnet bei der Materialstrukturanalyse verschiedener anorganischer chemischer Zusammensetzungen gute Ergebnisse liefert.Bei der Simulationskristallgröße von 100 Å ist die Leistung etwas besser, bleibt aber bei der anspruchsvolleren Simulationskristallgröße von 10 Å ausgezeichnet.Beispielsweise kann PXRDnet die Kristallform von Materialien wie Cs₂YCuCI₆ und SmMn₂SiC erfolgreich erfassen, und es kann auch die Symmetrie von Materialien wie Cs₂YCuCI₆ und BaSrMnWO₆ erfolgreich erfassen. Darüber hinaus kann PXRDnet selbst in einigen Extremfällen, wie etwa beim Versagen von Li₅Nb₂Cu₃O₁₀ oder Sb₂F₁₃, noch immer wertvolle Referenzen für Experimente liefern.
Die folgende Abbildung zeigt den Vergleich des realen PXRD-Musters, des ursprünglich von PXRDnet vorhergesagten Musters und des Musters nach der Rietveld-Verfeinerung. Dies zeigt den Grad der Übereinstimmung zwischen dem vorhergesagten Modell und den realen Daten und bestätigt die Notwendigkeit von Rietveld, wodurch die Vorhersagegenauigkeit des Modells effektiv verbessert werden kann. Beispielsweise betrug bei 100 Å die vorhergesagte Differenz für Sb₂F₁₃ 0,681, die nach der Verfeinerung (AI+Rietveld) auf 0,019 reduziert wurde.
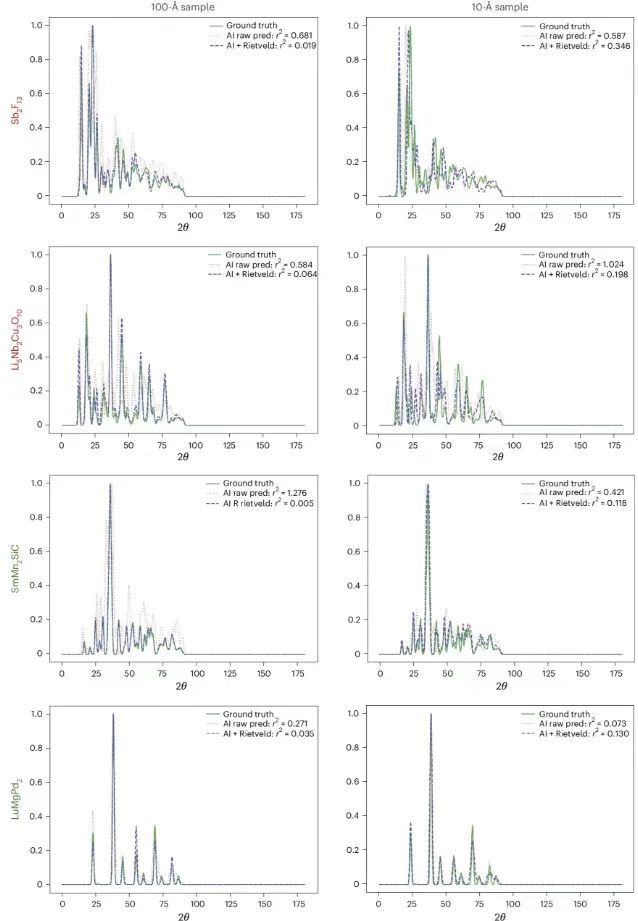
Vergleich des wahren PXRD-Musters, des ursprünglich von PXRDnet vorhergesagten Musters und des Musters nach der Rietveld-Verfeinerung
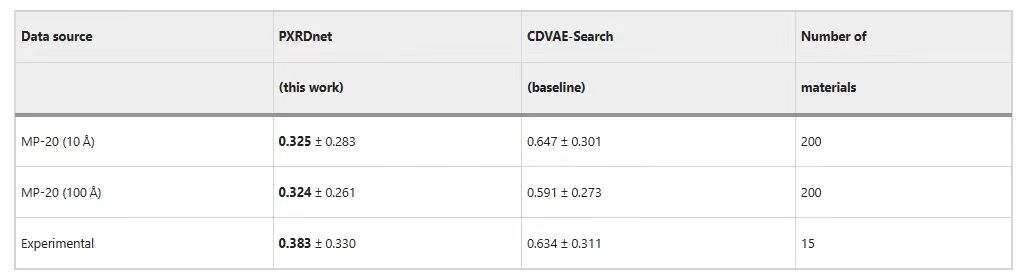
Die folgende Tabelle zeigt, dass PXRDnet die Materialien in MP-20 erfolgreich rekonstruieren kann.Im Vergleich zur CDVAE-Search-Basislinie sind die Vorhersageergebnisse von PXRDnet hervorragender.
Um die Ergebnisse weiter zu verbessern, führten die Forscher eine Rietveld-Verfeinerung an 20 gleichmäßig ausgewählten, von PXRDnet aufgelösten Strukturen durch und wählten für jede Struktur die zehn besten Kandidateneingaben aus. Wie in der Abbildung unten gezeigt.
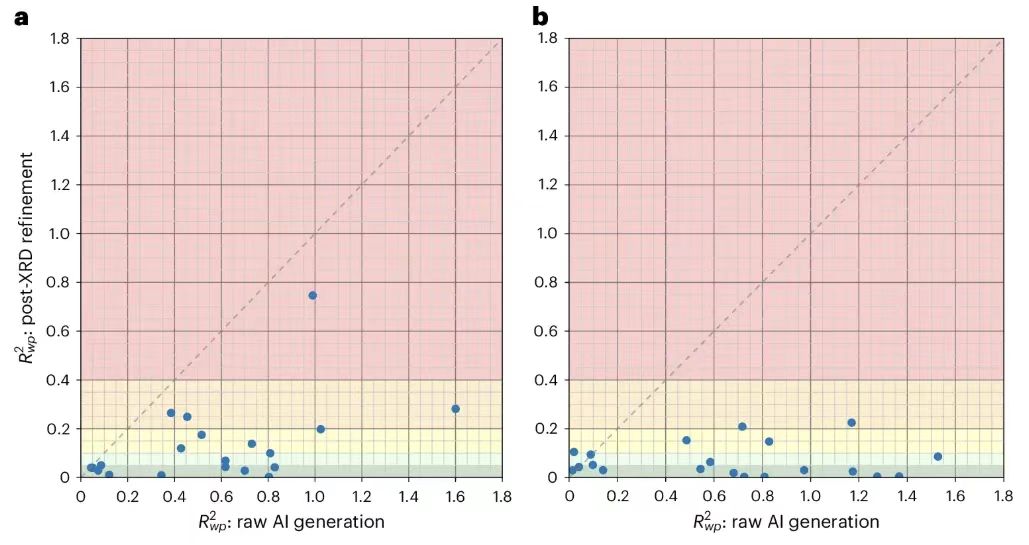
Die Ergebnisse zeigen, dassDie Rietveld-Verfeinerung war besonders effektiv bei den 100-Å-Tests, die schärfere Bragg-Peaks aufweisen, wobei 18 der 20 getesteten Strukturen unter 20% und 15 unter 10% lagen.Dies zeigt, dass PXRDnet trotz einiger kleinerer Probleme immer noch in der Lage ist, konsistent Ergebnisse auszugeben, die nahe an der wahren Struktur liegen, und dass mit entsprechendem menschlichen Eingriff in jedem Fall die richtige Struktur erzielt werden kann.
Abschließend überprüften die Forscher die Leistung des PXRD-Tests experimentell mit Daten aus der IUCr-Datenbank. Wie in der Abbildung unten gezeigt.
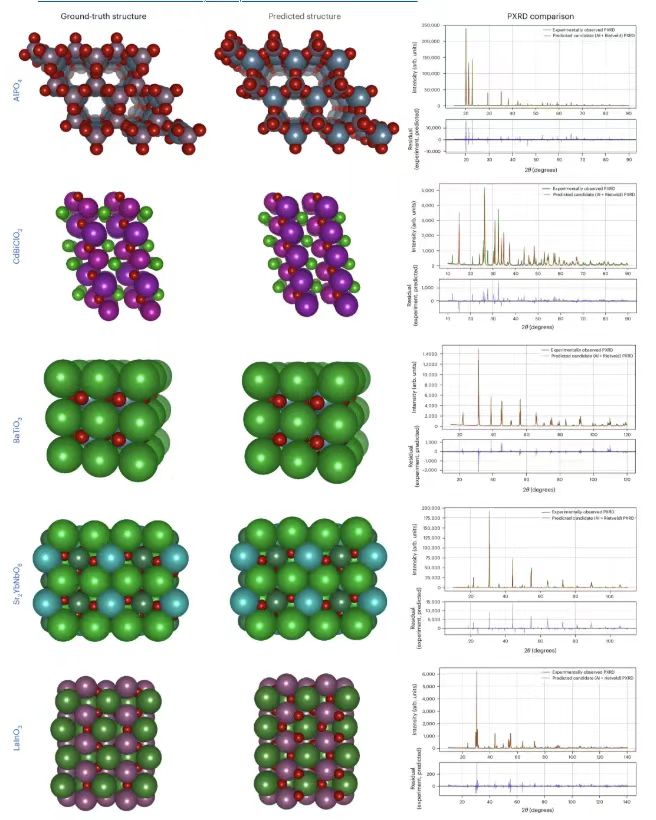
Die Spalte ganz links zeigt die Benchmark-Strukturen, basierend auf den experimentell beobachteten PXRD-Mustern aus der IUCr-Datenbank, die mittlere zeigt die von PXRDnet vorhergesagten Strukturen und die rechte Seite zeigt den Vergleich zwischen dem mit TOPAS (v.7) simulierten PXRD und dem tatsächlich experimentell beobachteten PXRD.Die Ergebnisse zeigen, dass PXRDnet die Lücke zwischen Simulation und Realität überwindet und dass seine Ergebnisse hinsichtlich visueller Analyse und quantitativer Messwerte mit denen aus simulierten Daten vergleichbar sind. Dies verdeutlicht das Potenzial des vorgeschlagenen Modells für die Anwendung in realen Szenarien.
KI und Materialwissenschaft lösen gemeinsam jahrhundertealte Probleme
Die Einführung von PXRDnet löste ein jahrhundertealtes Problem in der Materialwissenschaftsgemeinschaft. Wie in der Abhandlung dargelegt, ist die Methode, wie jede Strukturlösung, nicht zu 100 % erfolgreich, sie bietet jedoch eine mögliche Methode zur Erforschung der Strukturaufklärung und öffnet somit weitere Türen zum Erfolg.
Natürlich kam der Erfolg von PXRDnet nicht über Nacht, sondern war das Ergebnis kontinuierlicher Forschung, bei der das Unternehmen auf den Schultern von Giganten stand. An der Schnittstelle zwischen künstlicher Intelligenz und Nanomaterialien arbeiten unzählige Wissenschaftler ständig an Durchbrüchen.
Ein Beispiel hierfür ist die vom MIT, der Stanford University und anderen Teams veröffentlichte Forschung zum Thema „Kristallstrukturbestimmung anhand von Pulverbeugungsmustern mit generativem maschinellem Lernen“.Hier präsentieren wir ein bahnbrechendes generatives maschinelles Lernmodell, das Kristallstrukturen aus echten experimentellen PXRD-Daten lösen kann.In den Experimenten sagten die Forscher die Strukturen von 134 experimentellen Mustern aus der RRUFF-Datenbank und Tausenden simulierten Mustern aus dem Materials Project voraus, wobei die Modellübereinstimmungsraten den neuesten Stand der Technik von 42% bzw. 67% erreichten.
Papieradresse:
https://pubs.acs.org/doi/10.1021/jacs.4c10244
Darüber hinaus haben auch Teams der Chinesischen Akademie der Wissenschaften, der Shanghai Jiaotong University, der Tsinghua University und der Renmin University of China entsprechende Forschungsergebnisse veröffentlicht.Wir schlagen ein End-to-End-Neuralnetzwerk, PXRDGen, vor, das die Kristallstruktur bestimmen kann, indem es die strukturelle Verteilung experimentell stabiler Kristalle und ihrer PXRD-Muster lernt.Die atomare Präzisionsstruktur wurde aus PXRD-Daten extrahiert. Das Modell verfügt über einen vortrainierten XRD-Encoder, einen auf Diffusion/Fluss basierenden Strukturgenerator und ein Rietveld-Verfeinerungsmodul und kann in nur wenigen Sekunden eine präzise Strukturauflösung erreichen. Die entsprechende Forschung wurde unter dem Titel „Powder Diffraction Crystal Structure Determination Using Generative Models“ veröffentlicht.
Papieradresse:
https://arxiv.org/abs/2409.04727
Zusammenfassend lässt sich sagen, dass die Erforschung von PXRDnet und anderen Methoden es der Materialwissenschaftsgemeinschaft ermöglicht hat, von traditionellen Methoden zur übergreifenden Integration von künstlicher Intelligenz und Materialwissenschaft überzugehen. Dabei wurden nicht nur wesentliche Durchbrüche erzielt und Probleme gelöst, mit denen die Materialwissenschaftsgemeinschaft konfrontiert war, sondern auch neue Ideen und Methoden für nachfolgende Forschungen bereitgestellt und so der zukünftigen Entwicklung der Materialwissenschaft neue Vitalität verliehen.








