Command Palette
Search for a command to run...
Die Neueste Forschung Von David Bakers Team Nutzt Modelle Zur Proteinsequenzgenerierung, Um Überlappendes Gendesign Mit Einer Sehr Hohen Erfolgsrate Zu Erreichen
Im Jahr 1977 entdeckte der britische Biochemiker Frederick Sanger bei der Analyse des Genoms des Bakteriophagen ΦX174 erstmals ein Phänomen, das die kognitiven Fähigkeiten untergrub: Die Gesamtlänge der von diesem 5,4 kb großen DNA-Molekül kodierten Proteine überschritt die physikalische Kapazitätsgrenze bei weitem. Die Sequenzierungsergebnisse zeigten, dassZwei Genpaare teilen sich dieselbe DNA-Region über unterschiedliche Leserahmen – dieses Phänomen wird als überlappende Gene (OLG) bezeichnet und kommt in der viralen Welt äußerst häufig vor.Beispielsweise ist im 3,2 kb großen Genom des Hepatitis B-Virus die Region 50% von mehreren Paaren überlappender Gene bedeckt, und mehr als die Hälfte der bekannten Viren enthält mindestens ein OLG.
Hinter diesem wenig intuitiven Genomdesign verbirgt sich die Überlebensweisheit des Virus: Wenn Viren in Wirtszellen um begrenzten Platz konkurrieren, verwendet OLG die Strategie der „Genstapelung“, um einem einzelnen Nukleotid die gleichzeitige Beteiligung an der Kodierung zweier Codons zu ermöglichen und so eine funktionelle Überlagerung in einer kompakten Sequenz zu erreichen. Die Entdeckung des Sanger-Teams löste entsprechende Forschungen aus. Nachfolgende Studien haben gezeigt, dass die von OLG kodierten Proteine häufig eine hohe Sequenzdegeneration aufweisen und ihre Aminosäuresequenztoleranz es ermöglicht, dass zwei funktionelle Proteine auf derselben DNA-Kette koexistieren. Noch wichtiger ist, dass selbst Proteine, die eine klare dreidimensionale Struktur bilden müssen, durch Sequenzanordnung eine Faltungskompatibilität in verschiedenen Leserahmen erreichen können.
Die Kernfrage bleibt jedoch immer bestehen: Kann die Degeneration von Aminosäuresequenzen im Rahmen des standardmäßigen genetischen Codes die Faltung beliebiger funktioneller Proteinpaare in überlappenden Strukturen unterstützen? Wenn bei Nukleotiden eine duale Kodierung berücksichtigt werden muss, ist dann der Sequenzraum für die Proteinfaltung stark eingeschränkt?
Das Team von David Baker an der University of Washington verwendete vor Kurzem fortschrittliche generative Modelle, um synthetische OLG-Designforschung zu betreiben und deren Machbarkeit aus technischer Sicht zu überprüfen.Das Forschungsteam entwarf überlappende Sequenzen für zwei Proteinfamilien, um hochgeordnete, neu entworfene Proteinstrukturen zu kodieren. Sowohl die Computersimulation als auch die experimentelle Überprüfung zeigten eine sehr hohe Erfolgsrate: Unter Überlappungsbeschränkungen können alternative Leserahmen nicht nur eine klare dreidimensionale Faltung ermöglichen, sondern auch ihre strukturelle Stabilität und funktionelle Integrität sind mit denen nicht überlappender Sequenzen vergleichbar.
Die entsprechenden Forschungsergebnisse wurden als Preprint auf bioRxiv unter dem Titel „Design of overlapping genes using deep generative models of protein sequences“ veröffentlicht.

Papieradresse:
https://doi.org/10.1101/2025.05.06.652464
Das Open-Source-Projekt „awesome-ai4s“ vereint mehr als 100 AI4S-Papierinterpretationen und stellt umfangreiche Datensätze und Tools bereit:
https://github.com/hyperai/awesome-ai4s
Datensatz: Integration multidimensionaler Datenressourcen und Analysemethoden
Um die Plastizität des genetischen Codes und seine Anwendung im Proteindesign zu analysieren, integriert diese Studie mehrdimensionale Datenressourcen und Analysemethoden, um eine vollständige Forschungskette vom theoretischen Design bis zur experimentellen Verifizierung aufzubauen.
In Studien zur Randomisierung des genetischen CodesDie Studie generierte 1.000 alternative Codon-Kombinationen basierend auf Aminosäure-Permutationen und Codon-Shuffler-Strategien.Dieser Datensatz gewährleistet durch klares Algorithmusdesign die Vielfalt und Einheitlichkeit der Proben und bietet einen statistischen Maßstab für die Bewertung der funktionellen Auswirkungen von Codon-Umlagerungen.
Gleichzeitig wurden im Rahmen der Studie drei repräsentative Zielproteine der Sekundärstruktur ausgewählt und neun Paarkombinationen erstellt, wodurch eine Standardisierung der Versuchsbedingungen unter der Prämisse der Kontrolle von Variablen erreicht und die Korrelationsanalyse zwischen der Variation des genetischen Codes und der Funktion der Proteinstruktur effektiv verknüpft wurde.
In der Phase der Proteindomänensequenzanalyse extrahierte die Studie Startsequenzen aus der Pfam 37.0-Datenbank, wählte zufällig Unterregionen mit einer Länge von 100 Aminosäuren aus und verwendete das Markov-Modell, um synthetische Proteinsequenzen zu generieren, die die K-Mer-Verteilung beibehielten.Diese Methode kombiniert bioinformatisches Screening und statistische Modellierung, wodurch nicht nur die Sequenzmerkmale natürlicher Proteine erhalten bleiben, sondern durch die Einführung kontrollierbarer Zufallsvariablen auch Kontrollproben erstellt werden.Es bietet einen innovativen Datensatz, der natürliche Eigenschaften mit künstlich gestalteten Merkmalen für die anschließende Analyse kombiniert.
Bei der Einbettungsanalyse des Proteinsprachenmodells extrahierten die Forscher die verborgenen Schichtmerkmale von ESM2, ESM3 und ProstT5 und projizierten sie nach der Positionsmittelung mithilfe des UMAP-Algorithmus in den zweidimensionalen Raum. Durch die genaue Einstellung von Parametern wie n_neighbors = 15 werden hochdimensionale Sequenzmerkmale in intuitive topologische Karten umgewandelt.Unter Beibehaltung der Sequenzähnlichkeitsstruktur bietet es einen einheitlichen Visualisierungsrahmen für modellübergreifende Vergleiche.Es demonstriert die hochmoderne Kombination aus Computerbiologie und Datenvisualisierung.
Während der experimentellen VerifizierungsphaseDie Forscher klonten und rekombinierten 192 überlappende Gene, um 384 Proteinvarianten mit verschobenem Leseraster zu erzeugen.Die Schlüsselparameter wurden im Experiment streng kontrolliert: 20 Stunden Kultur bei 37 °C stellten die Stabilität des E. coli-Expressionssystems sicher und ein Renaturierungsschema mit einem 6M Guanidinhydrochlorid-Gradienten stellte die korrekte Faltung des Einschlusskörperproteins sicher. Diese quantitative Kontrolle des gesamten Prozesses vom Moleküldesign bis zur Reinigung und Charakterisierung verbessert nicht nur die Reproduzierbarkeit von Forschungsergebnissen, sondern bietet auch ein standardisiertes experimentelles Paradigma für das Protein-Engineering.
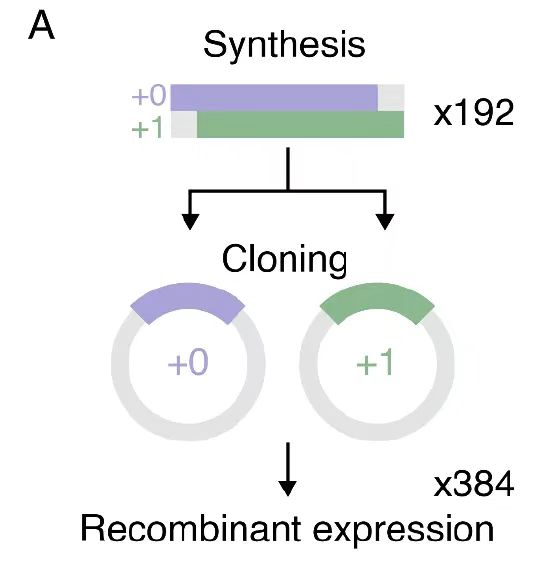
OLG-Design basierend auf einem generativen Modell: eine mit mehreren Frameworks kompatible Methode zur Optimierung der Sequenzsynchronisation
Im Rahmen dieser Studie wurde ein Computeralgorithmus entwickelt, der das Problem der Sequenzraumbeschränkungen, die durch die gegenseitige Abhängigkeit codierender Frames beim überlappenden Gendesign (OLG) verursacht werden, wirksam angeht und eine gleichzeitige Optimierung der Anpassungsfähigkeit zweier Proteinsequenzen ermöglicht.
Auf der Ebene des Algorithmus-Designs integrierte die Studie generative Modelle wie EvoDiff-MSA und ProteinMPNN.Ersteres basiert auf der MSA-Transformer-Architektur und kann durch autoregressives Diffusionszieltraining Designsequenzen basierend auf der Multiple Sequence Alignment (MSA) des Zielproteins generieren. Letzteres kann als strukturelles bedingtes Generierungsmodell entsprechende Proteinsequenzen entwerfen, wenn eine dreidimensionale Struktur gegeben ist. Beide Modelle verwendeten Position-für-Position-Maskierung und eingeschränkte Sampling-Strategien, um überlappende Sequenzbibliotheken zu generieren, die eine Vielzahl von Offsets und Frame-Anordnungen abdecken.
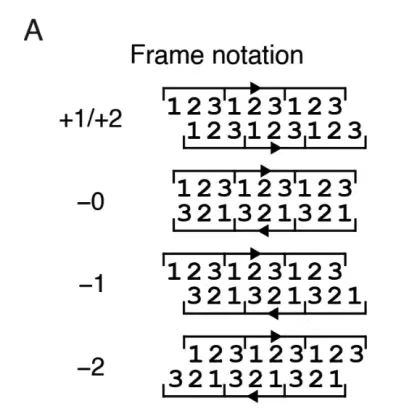
Wie in Abbildung A unten dargestellt, wurde in dieser Studie eine iterative Einzelbild-Sampling-Strategie für die Phasenbeschränkungen von fünf variablen Leserahmen (+1, +2, -0, -1 und -2) vorgeschlagen.
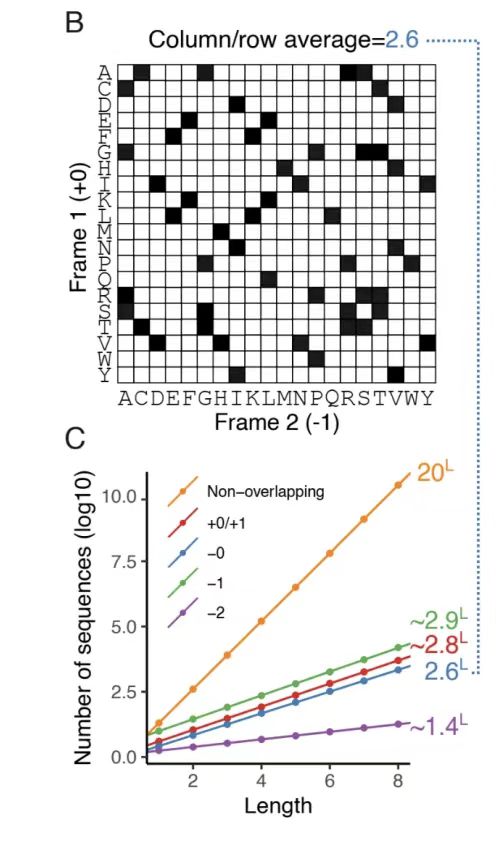
Wie in Abbildung B unten gezeigt, wurde durch die Analyse der Aminosäurekompatibilitätsmatrix des -0-Frameworks festgestellt, dass an einer einzigen Position im Referenzframework durchschnittlich 2,6 kompatible Aminosäuren zur Auswahl stehen, wodurch 52ⁿ (n ist die Sequenzlänge) potenziell überlappende Sequenzpaare entstehen, was den durch die Degeneration des genetischen Codes bedingten Gestaltungsspielraum verdeutlicht. Die Freiheitsgrade anderer Frameworks wurden mithilfe der Monte-Carlo-Approximation quantifiziert, wie in Abbildung C unten dargestellt. Die Ergebnisse zeigen, dass die +1- und -1-Frameworks höhere Freiheitsgrade aufweisen (etwa 2,8 bzw. 2,9), während das -2-Framework aufgrund der geringen Effizienz der Codon-Degeneration deutlich eingeschränkte Freiheitsgrade (etwa 1,4) aufweist.
Schließlich scannt der Algorithmus, wie in Abbildung D unten gezeigt, systematisch die Sequenzpositionen (Scan-Reihenfolge) und aktualisiert die gemeinsame Wahrscheinlichkeitsmatrix in jedem Scan dynamisch in Kombination mit den Einschränkungen der benachbarten Aminosäuren.Nach mehreren Iterationsrunden wird sichergestellt, dass die generierten überlappenden Sequenzpaare die Kompatibilität des Frameworks erfüllen.Diese Strategie kann auf komplexe Frameworks mit Phasenversatz erweitert werden, wobei die Designqualität durch die Beeinflussung der Scanreihenfolge und die Bereitstellung wichtiger Einschränkungen für die iterative Dekodierung des generativen Modells optimiert wird.
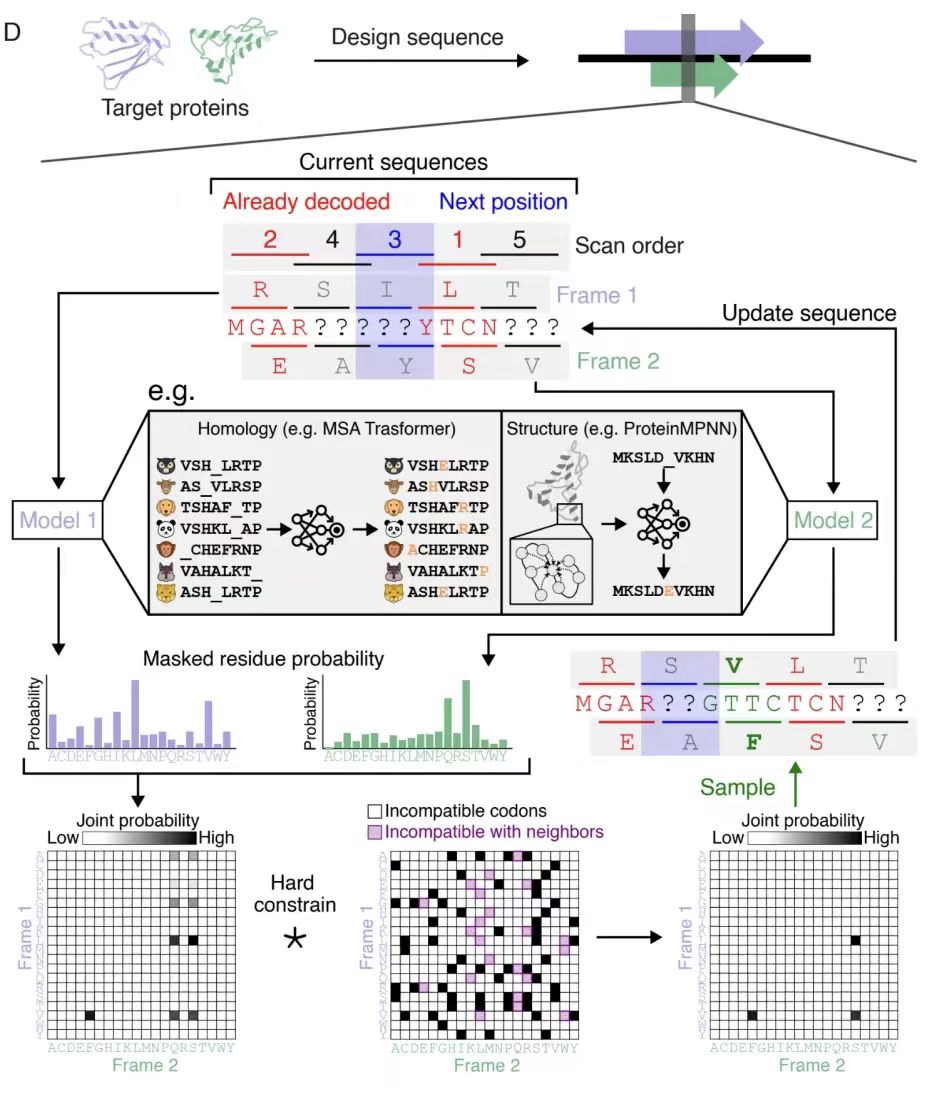
Jenseits der natürlichen Template-Beschränkung: Effiziente Erzeugung synthetischer OLGs beliebiger Proteinpaare
Das experimentelle Design deckt mehrere Richtungen ab, darunter die auf Homologie basierende Bewertung des OLG-Designs, die Analyse überlappender Machbarkeiten hochgeordneter Protein-Hauptkettenstrukturen, Studien zur evolutionären Zugänglichkeit von OLG-Sequenzen und die experimentelle Verifizierung.
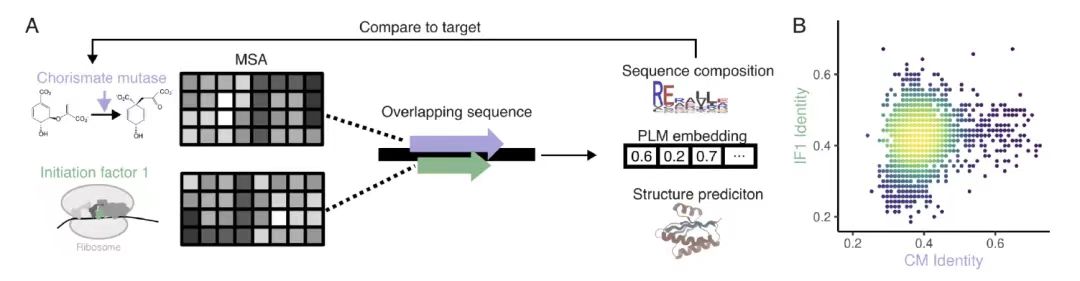
In der homologiebasierten OLG-DesignbewertungWie in Abbildung A unten gezeigt, wählte das Forschungsteam bakterielle Shikimatmutase (CM) und Translationsinitiationsfaktor 1 (IF1) als Ziele, verwendete das EvoDiff-MSA-Generierungsmodell und nutzte die multiple Sequenzalignmentierung (MSA) als bedingten Kontext, um durch Position-für-Position-Maskierung und eingeschränkte Stichprobennahme 3.307 vollständig überlappende Sequenzdesigns zu generieren.
Wie in Abbildung B unten gezeigt, beträgt die Homologie zwischen der entworfenen Sequenz und der natürlichen Sequenz zwar nur 38,9% (CM) und 42,3% (IF1),Die Einbettungsanalyse des Proteinsprachenmodells zeigt jedoch, dass seine Verteilung im zweidimensionalen Raum in hohem Maße mit der natürlichen Sequenz übereinstimmt.Dies deutet darauf hin, dass es sich bei den entworfenen Sequenzen um glaubwürdige Mitglieder der Zielproteinfamilie handelt, und bestätigt die Entwurfsfähigkeiten des Algorithmus für natürliche Proteinfamilien.
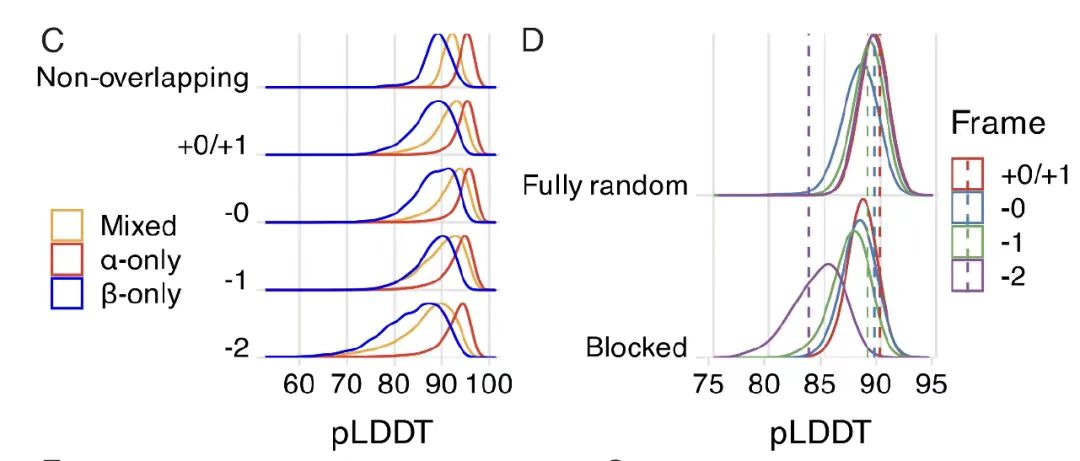
Bei der Untersuchung der Machbarkeit überlappender hochgeordneter Protein-RückgratstrukturenWie in Abbildung A unten dargestellt, verwendeten die Forscher das strukturbedingte Generierungsmodell ProteinMPNN, um 56.250 überlappende Designs und 33.000 nicht überlappende Designs für 15 de novo generierte Hauptkettenstrukturen (die die Kategorien α, β und gemischte Faltung abdecken) zu generieren. Wie in Abbildung B unten dargestellt, zeigen die AlphaFold2-Evaluierungsdaten, dassDer durchschnittliche pLDDT-Wert für das überlappende Design betrug 90,2 und lag damit nahe am Wert von 92,0 für das nicht überlappende Design.
Weitere Analysen ergaben, dass, wie in Abbildung CD unten gezeigt, nur der -2-Frame aufgrund der geringen Effizienz der Codon-Degeneration eine schlechte Leistung zeigte. Die randomisierte genetische Codeanalyse zeigte, dass der natürliche genetische Code (SGC) einen erheblichen Vorteil bei der Kodierung von OLG hat, mit Ausnahme des -2-Frames gute Leistungen erbringt und eine Zusammensetzungspräferenz für stark degenerierte Aminosäuren aufweist.Der Mechanismus, durch den die SGC-Struktur die Durchführbarkeit überlappender Sequenzen beeinflusst, wurde aufgedeckt.
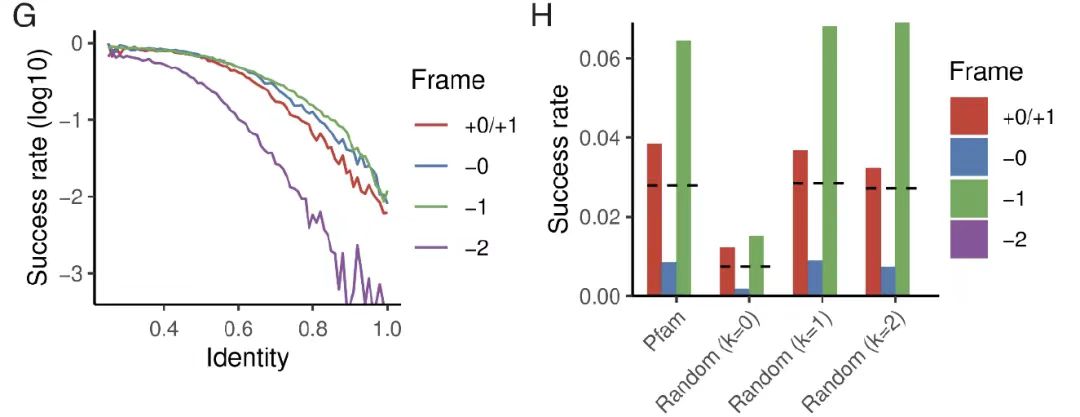
In Studien zur evolutionären ZugänglichkeitDas Forschungsteam begann mit einer Samenproteinsequenz mit einer festen Anzahl von Mutationen. Wie in der folgenden Abbildung GF gezeigt,Die Studie ergab, dass selbst unter der extremen Bedingung einer Nullmutation etwa 1%-Designs immer noch eine hohe strukturelle Stabilität erreichen können (pLDDT > 85, TM > 0,7).Bei Verwendung natürlicher Pfam-Sequenzen als Eltern erhöhte sich die Erfolgsrate auf 3%, und dieses Ergebnis stimmte mit zufälligen Sequenzen überein, die die Zusammensetzungsabweichung erster Ordnung beibehielten. Dies zeigt deutlich, dass hochoptimierte natürliche Proteine neue Proteine in alternativen Strukturen ohne größere Sequenzänderungen aufnehmen können, was die Machbarkeit von OLG auf evolutionärer Ebene bestätigt.
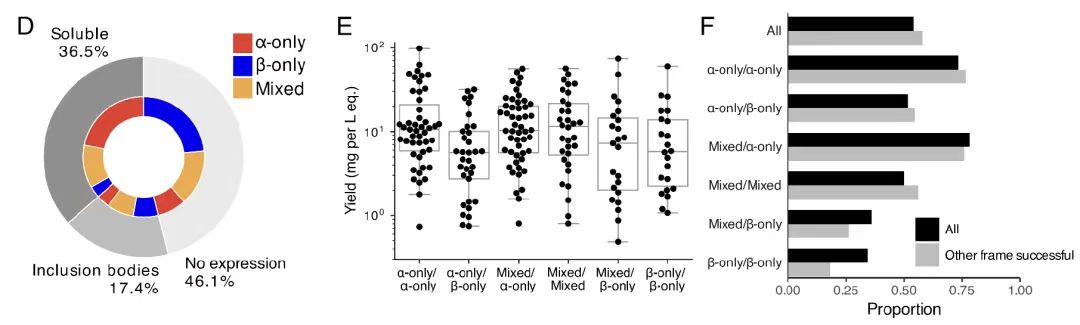
Im letzten Teil der experimentellen Verifizierung führte das Forschungsteam eine rekombinante Expression und strukturelle Charakterisierung an 192 überlappenden Sequenzen durch. Die Ergebnisse zeigen, dass, wie in Abbildung B dargestellt,Einzelne Proteine von 54% wurden erfolgreich exprimiert und die meisten wiesen die erwarteten Sekundärstrukturen und eine hohe thermische Stabilität auf.
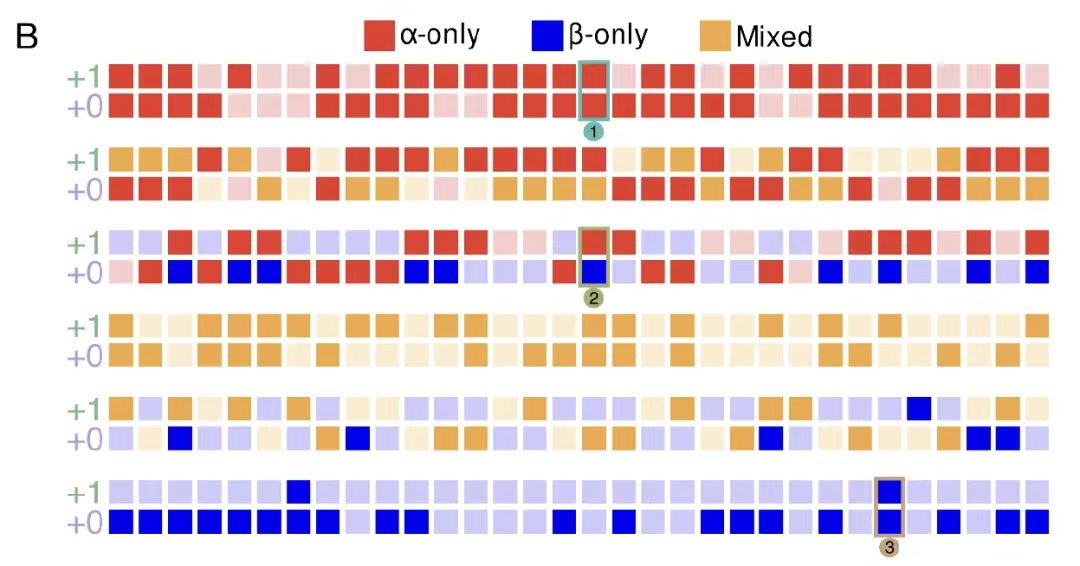
Darüber hinaus variierte die Erfolgsrate, wie in den Abbildungen DF unten gezeigt, je nach Sekundärstrukturgehalt des Proteins, wobei α-helikale Proteine die höchste Erfolgsrate aufwiesen. Darüber hinaus wurden überlappende Paare von 31% erfolgreich gereinigt, und der Erfolg eines Frameworks hatte keinen Einfluss auf den Erfolg des anderen.Diese Ergebnisse untermauern die hohe Machbarkeit und experimentelle Validierungsrate der OLG-Sequenzen und demonstrieren die Wirksamkeit des Algorithmus beim Entwurf funktioneller und strukturell stabiler überlappender Proteine.
Grenzforschung im Bereich der synthetischen Biologie, OLG-Engineering-Anwendungen vertiefen sich schrittweise
Auf dem Gebiet der synthetischen Biologie beschäftigen sich Forschungsteams und Unternehmen in vielen Teilen der Welt eingehend mit der technischen Anwendung überlappender Gene (OLGs).
Beispielsweise hat die Forschungsgruppe von Zhu Ting an der Tsinghua-Universität bedeutende Fortschritte bei der Untersuchung von Spiegelbiologiesystemen erzielt und die vollständige chemische Synthese der Spiegel-Pfu-DNA-Polymerase erfolgreich durchgeführt.Dadurch wird nicht nur die Zusammensetzung von Spiegel-DNA mit einer Länge von mehreren Kilobasen möglich, sondern auch die Entwicklung einer auf Spiegel-DNA basierenden Informationsspeichertechnologie ermöglicht.Diese Technologie nutzt die Kodierungsstrategie von Spiegelgenen, um eine neue Idee für die bidirektionale funktionelle Überlagerung von OLG zu liefern. Wenn die Doppelhelixstruktur der Spiegel-DNA sowohl natürliche als auch gespiegelte genetische Informationen enthält, wird die Nutzung des Sequenzraums erheblich verbessert, was eine wichtige Grundlage für das kompakte Design künstlicher Genome bietet.
* Link zum Artikel:https://www.nature.com/articles/s41587-021-00969-6
Darüber hinaus hat das Team von Christopher Voigt am Massachusetts Institute of Technology eine synthetische Biologieplattform entwickelt, die auf dem Design von Genschaltkreisen basiert. Durch die Rekonstruktion der Regulationslogik prokaryotischer Gencluster gelang ihnen die modulare Zusammenstellung von Stoffwechselwegen. Dieser technische Weg ist eng an der Designphilosophie von OLG ausgerichtet.Wenn mehrere funktionelle Gene durch überlappende Sequenzen ein kompaktes genetisches Modul bilden, kann dies nicht nur die Redundanz des Genoms reduzieren, sondern durch koordinierte Expression auch die Stabilität des Systems verbessern.Beispielsweise übernahm der vom Team entwickelte künstliche Stickstofffixierungs-Gencluster die OLG-Strategie, um die codierenden Sequenzen mehrerer Schlüsselenzyme in dieselbe DNA-Region zu komprimieren. Dadurch wurde die metabolische Belastung der Wirtszellen deutlich reduziert und gleichzeitig die katalytische Effizienz sichergestellt.
* Link zum Artikel:https://www.nature.com/articles/s41467-022-33272-2
Es ist erwähnenswert, dass diese Studien nicht nur die weit verbreitete Existenz von OLG in der natürlichen Evolution belegen, sondern auch seine biophysikalische Machbarkeit mit technischen Mitteln bestätigen. In der in diesem Artikel vorgestellten Studie verwendete das Team von David Baker ein Deep-Learning-Modell zum Entwurf synthetischer OLGs, die in Computersimulationen eine strukturelle Stabilität zeigten, die mit der natürlicher Sequenzen vergleichbar ist. Die hohe Erfolgsrate der experimentellen Verifizierung belegt zusätzlich die biologische Kompatibilität der überlappenden Kodierung. Dieser vollständige geschlossene Kreislauf von der Grundlagenforschung bis zur angewandten Transformation verändert die Designlogik der synthetischen Biologie und dürfte in vielen Bereichen, wie etwa der innovativen Arzneimittelentwicklung, der Präzisionsdiagnose und der Zelltherapie, zu neuen Durchbrüchen führen.
Quellen:
1.https://www.tsinghua.edu.cn/info/1181/86148.htm
2.https://tech.huanqiu.com/article/9CaKrnJUV0x
3.https://news.bioon.com/article/4161e88572ad.html








