Command Palette
Search for a command to run...
Das Für Die ICLR 2025 Oral Ausgewählte Team Von Zhou Hao Vom Tsinghua AIR Schlug Ein Neues Paradigma Für Das Protein-Vortraining Vor, Um Die Evolution Der Proteinfamilie Zu Entschlüsseln

Die AIR GenSI-Forschungsgruppe der Tsinghua-Universität und die School of Pharmacy der Tsinghua-Universität haben gemeinsam ein Tool für proteinfamilienspezifische generative Modellierung vorgeschlagen – ProfileBFN (Profile Bayesian Flow Network). ProfileBFN erweitert das diskrete Bayes-Flussnetzwerk aus der Perspektive von Multiple Sequence Alignment (MSA)-Profilen, um ein effizientes Proteinfamiliendesign zu erreichen. Die empirischen Ergebnisse zeigen, dassWährend ProfileBFN vielfältige und neuartige Familienproteine erzeugt, ist es in der Lage, die strukturellen Merkmale der Familie genau zu erfassen.

Die entsprechenden Ergebnisse trugen den Titel „Steering Protein Family Design through Profile Bayesian Flow“ und wurden als mündlicher Vortrag bei ICLR 2025 ausgewählt. Gleichzeitig wurde eine weitere Errungenschaft des Teams, CrysBFN, auch für ICLR 2025 Spotlight ausgewählt. Der Titel der Forschungsarbeit lautet „Ein periodischer Bayes-Fluss zur Materialerzeugung“.
In der letzten Sitzung schlug das Team das geometrische Bayesianische Strömungsnetzwerk GeoBFN vor und die zugehörigen Ergebnisse wurden für die mündliche ICLR 2024 unter dem Titel „Vereinheitlichte generative Modellierung von 3D-Molekülen mit Bayesianischen Strömungsnetzwerken“ ausgewählt.

Link zum Artikel:
Das Open-Source-Projekt „awesome-ai4s“ vereint mehr als 200 AI4S-Papierinterpretationen und stellt umfangreiche Datensätze und Tools bereit:
https://github.com/hyperai/awesome-ai4s
Multiple Sequenzalignment: der Grundstein für die Vorhersage von Proteinstrukturen
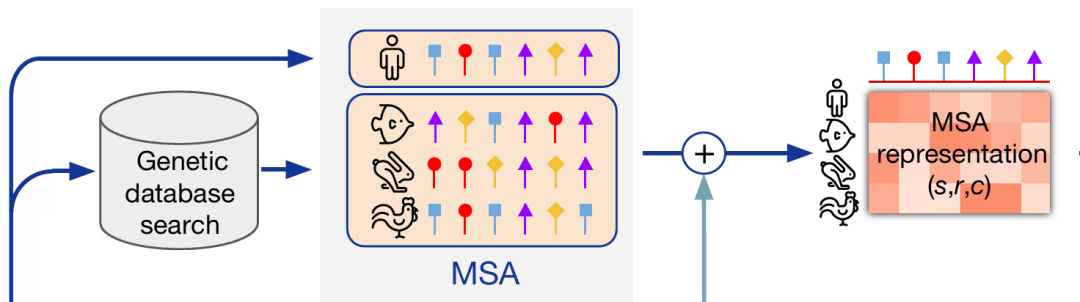
Unter Multiple Sequence Alignment (MSA) versteht man den Prozess der Ausrichtung von drei oder mehr biologischen Sequenzen (DNA, RNA oder Protein). Durch die multiple Sequenzalignmentierung können aufgrund funktioneller, struktureller oder evolutionärer Beziehungen ähnliche Regionen entdeckt und identifiziert werden, wodurch eine umfassendere Perspektive auf die Beziehungen zwischen biologischen Makromolekülen gewonnen wird.
In den letzten Jahren ist die Nutzung von MSA-Informationen zu einem wichtigen Teil des Proteindesigns geworden. In Meilensteinarbeiten wie AlphaFold und ESM gibt es spezielle Module, die MSA-Informationen kodieren:
Es gibt viele Abfolgen für den Erfolg und viele Abfolgen für den Misserfolg.
MSA ist eine wahre Fundgrube an evolutionären Informationen, doch die bestehenden Modelle scheinen ihre Fähigkeit, diese Informationen zutage zu fördern, zu überschätzen. Mit der Entwicklung der Technologie hat die MSA-Tiefe der tiefen generativen Modelleingaben weiter zugenommen, jedoch ist dabei ein Engpass aufgetreten, der die Kosteneffizienz der Hinzufügung von MSA-Informationen in Frage gestellt hat. Die eigentliche Ursache liegt darin, dass sowohl hinsichtlich der Quantität als auch der Qualität der MSAs erhebliche Unsicherheit besteht:
Als homologe Sequenzen bezeichnen Forscher Sequenzen, die bei der multiplen Sequenzalignment-Analyse einen gewissen Grad an Ähnlichkeit aufweisen. Was die Menge betrifft, kann es sein, dass für einige „verwaiste“ Proteine nicht mehr als 10 homologe Sequenzen vorhanden sind, während für andere Proteine mehr als 10.000 homologe Sequenzen durchsucht werden können, was bei großen Modellen zu großer Verwirrung führt und zu einer Verschwendung von Ressourcen und einer Beeinträchtigung der Effizienz führt.
Tatsächlich übersteigen die Wunder der Natur die menschliche Vorstellungskraft. Im Laufe von Milliarden Jahren der Evolution spiegeln konvergente Strukturen die Auswirkungen der natürlichen Selektion wider, während Mutationen neue Möglichkeiten für die Evolution bieten. Bei diesen besonderen Arten in besonderen Umgebungen bleiben häufig die ursprünglichen Erscheinungsinformationen am Anfang des Evolutionsbaums erhalten, was genau die Grundlage für die Ableitung der Koevolutionstheorie darstellt. Wenn homologe Sequenzen als Modelleingabe verwendet werden, werden diese Informationen zwangsläufig von einer großen Menge anderer irrelevanter Informationen überlagert, und es können nur Darstellungen mit hoher Wahrscheinlichkeit modelliert werden. Um dieses Problem zu lösen,ProfileBFN modelliert jeden Cluster homologer Sequenzen als einheitliche Darstellung, die unabhängig von der Anzahl ist.
Eine gute homologe Sequenz sollte so viele homologe Informationen wie möglich enthalten. Experimente zeigen, dass in den meisten Fällen durch die Verwendung einiger weniger homologer Sequenzen mit der größten Informationsentropie der gleiche Effekt erzielt werden kann wie durch die Verwendung von Hunderten homologer Sequenzen. Einige homologe Sequenzen unterscheiden sich nur in wenigen Aminosäuren, was dem Modell viele irreführende redundante Informationen liefert.
Profil: Der Grundstein des Protein-Sockelmodells der nächsten Generation
Wissenschaft basiert auf Entdeckungen.Die Innovation von ProfileBFN liegt in der Entdeckung der großen Informationsredundanz, die im ursprünglichen MSA vorhanden ist. Wenn 100 homologe Sequenzen nach der Methode der Informationsentropie sortiert werden, kann das Modell den gleichen Effekt erzielen, indem nur die ersten 20 zum Training verwendet werden. Dazu muss eine Brücke zwischen Einzelsequenz und Mehrfachsequenzen geschaffen werden, weshalb Profile angezeigt wird:
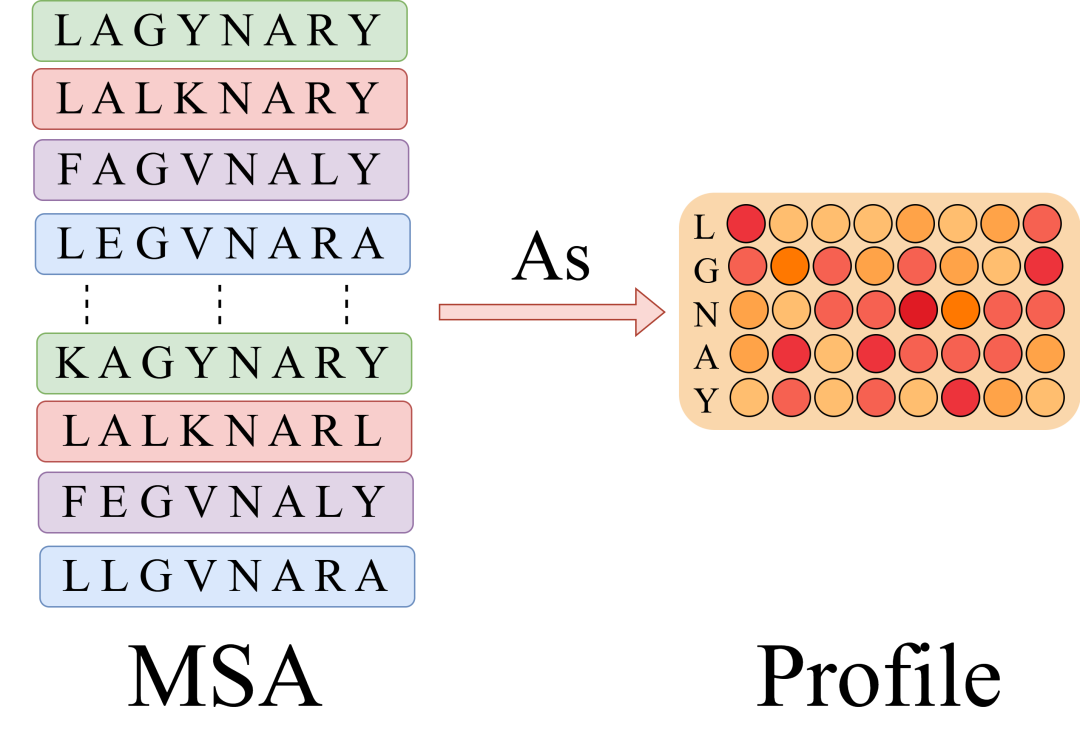
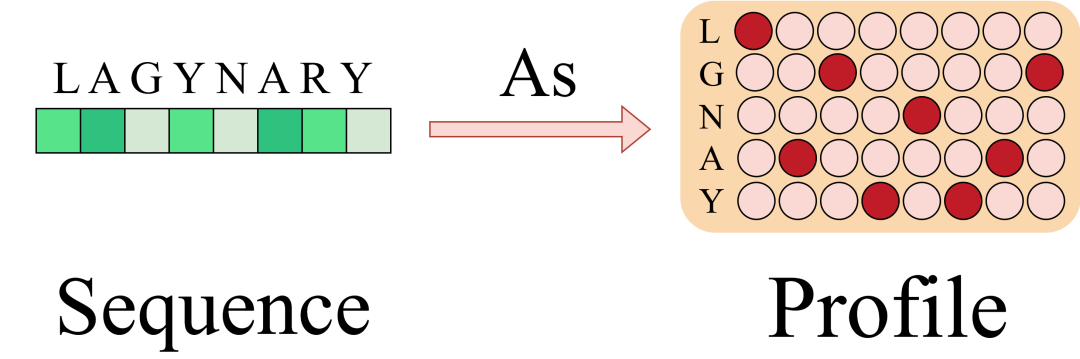
Um es intuitiv zu verstehen: „Profil“ ist eine spaltenweise Statistik der Anzahl der Aminosäurevorkommen in einer multiplen Sequenzausrichtung. Wenn darüber hinaus 1w-homologe Sequenzen mit einer Länge von jeweils 100 vorhanden sind, komprimiert Profile diese direkt von [10000,100] auf eine Liste von [20,100] (20 gemeinsame Aminosäuren), was den Rechenaufwand erheblich vereinfacht. Insbesondere kann auch eine einzelne Sequenz als spezielles Profil betrachtet werden, mit der Ausnahme, dass in jeder Spalte nur eine 1 steht.
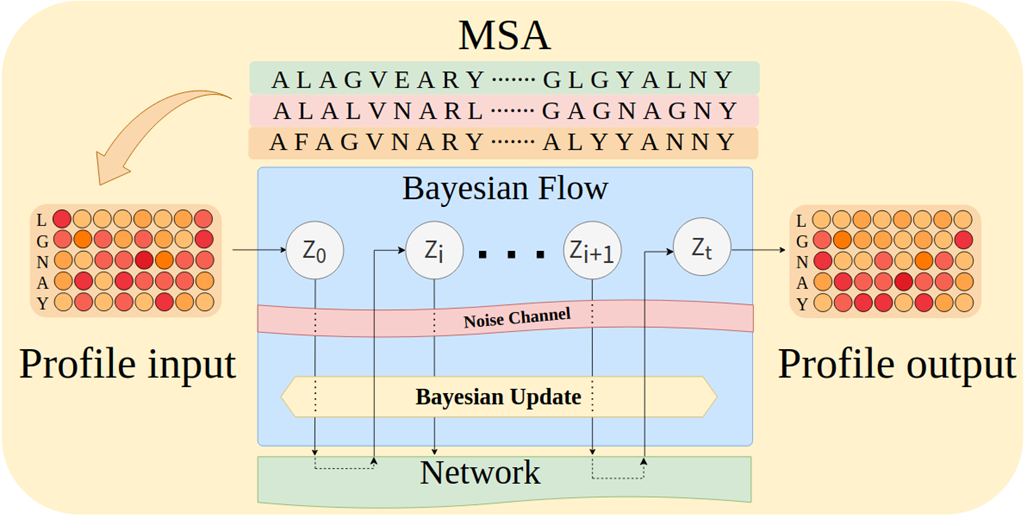
ProfileBFN stellte fest, dass die Komprimierung von MSA zu Profile nicht nur nicht den ursprünglich erwarteten schwerwiegenden Informationsverlust verursachte, sondern auch die Modellleistung erheblich verbesserte.Dies kann wie folgt verstanden werden: In der großen Welle des Bauprofils,Jede homologe Sequenz gibt Aufschluss über den Aminosäuretyp, der an dieser Position auftritt, wodurch kleinere Widersprüche verborgen und der Gesamttrend hervorgehoben werden.
ProfileBFNs unerwartet starke Leistung
Im Vergleich zur traditionellen Methode, die auf der Ausrichtung mehrerer Sequenzen basiert,ProfileBFN benötigt 10-mal weniger Daten und lernt 1,5-mal mehr Kontextinformationen über Proteinsequenzen.Die Wirkung tritt sofort ein!
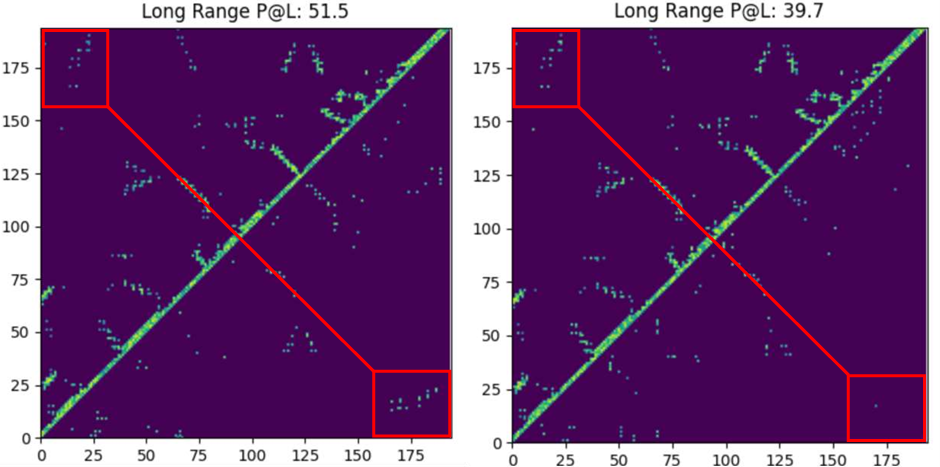
Nach der Untersuchung wurde bestätigt, dass ProfileBFN eine unterstützende Wirkung auf eine Vielzahl nachgelagerter Aufgaben hat:
* Enzymklassifizierung:Verbessern Sie die Funktionstreue und senken Sie die Screening-Kosten
* Lernen der Proteindarstellung:Unterstützung bei der Multitasking-Feature-Extraktion
* Vorhersage der Proteinstruktur:Verbessern Sie Homologieinformationen und Modellierungsgenauigkeit
* Antikörperproduktion:Hervorragender Migrationseffekt, genaue Vorhersage von Funktionsbereichen
Enzyme sind eine spezielle Klasse von Proteinen mit katalytischer Aktivität und ihre funktionelle Spezifität wird üblicherweise durch EC-Nummern (Enzyme Commission Numbers) beschrieben. Die Studie ergab, dass die neuen, durch ProfileBFN generierten Enzymkandidaten hinsichtlich der EC-Zahlen weitgehend mit dem Wildtyp-Enzym übereinstimmten, was bedeutet, dass die generierten Proteine ein hohes Maß an funktioneller Konsistenz beibehielten. Diese Funktion verringert den Aufwand für experimentelles Screening erheblich und verbessert die Erfolgsrate bei der Entwicklung neuer Enzyme.
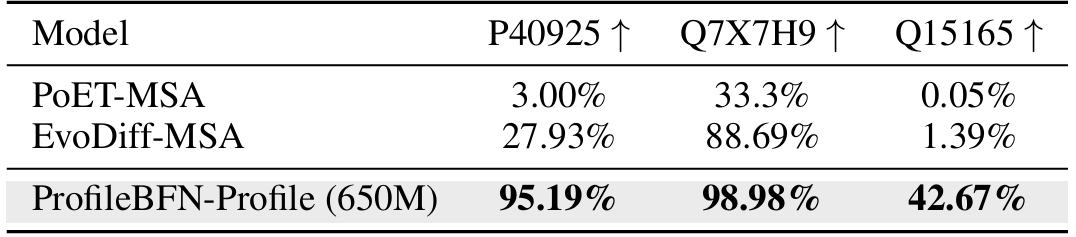
Während ProfileBFN Proteine generiert, erstellt es auch genaue Proteindarstellungen innerhalb des Modells. Die Forscher extrahierten diese Darstellungen,Es wurde anhand mehrerer Datensätze wie der thermischen Stabilität von Proteinen, der Proteininteraktion und der subzellulären Lokalisierung von Proteinen optimiert. Die Ergebnisse zeigten, dass die von ProfileBFN bereitgestellte Darstellung die Modellleistung bei nachgelagerten Aufgaben wie der Klassifizierung effektiv verbessern kann. Dies deutet darauf hin, dass es sich nicht nur um ein generatives Modell, sondern auch um ein leistungsstarkes Tool zum Erlernen von Merkmalen handelt.
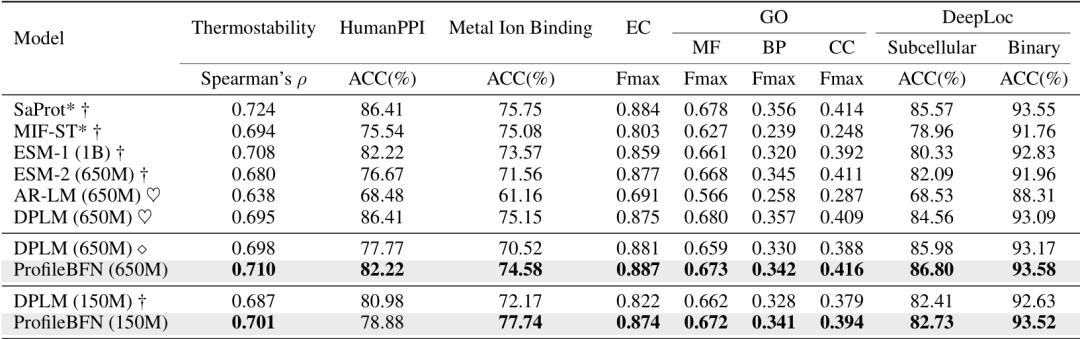
Die Vorhersage der Proteinstruktur ist ein wichtiges Thema in der Strukturbiologie.Insbesondere bei Orphan-Proteinen (d. h. Proteinen mit sehr wenigen homologen Proteinen) ist die Genauigkeit traditioneller Methoden stark eingeschränkt. Studien haben gezeigt, dass ProfileBFN als Verstärker von Homologieinformationen verwendet werden kann.Mit nur einer kleinen Menge an MSA-Daten werden mehr hochwertige homologe Proteine generiert, wodurch die Vorhersagegenauigkeit der Modelle der AlphaFold-Reihe verbessert wird. Diese Fähigkeit eröffnet ProfileBFN breite Anwendungsperspektiven im Bereich der Strukturbiologie.
Antikörper sind funktionelle Proteine, die spezifisch an Antigene binden können und in der Immun- und Pathologieforschung von großer Bedeutung sind. Um das Potenzial von ProfileBFN bei der Antikörpererzeugung zu erforschen,Die Forscher optimierten das Modell anhand der Antikörpersequenzdatenbank OAS (Observed Antibody Space).Die Ergebnisse zeigten, dass ProfileBFN bei der Erzeugung vielfältiger, hochwertiger Antikörpersequenzen gute Ergebnisse erzielte.
Die außergewöhnliche Wirkung von ProfileBFN beruht auf der Tatsache, dass diese neue Forschung ein Paradigma für die Generierung biologischer Sequenzen in der Post-MSA-Ära liefert:
* MSA nimmt nicht direkt als Input am Trainingsprozess teil und verursacht keinen zusätzlichen Trainingsaufwand
* In der Inferenzphase werden Einzelsequenz und MSA einheitlich modelliert
* Homologe Sequenzen sind sowohl Modell-Input als auch -Output
BFN nutzt Vorinformationen optimal
Da Profilinformationen sehr wichtig sind, sogar wichtiger als die ursprüngliche homologe Sequenz, wie sollten wir Profilinformationen verwenden? Das Bayesian Flow Network BFN passt perfekt zu Profile! Dies spiegelt sich in zwei Punkten wider:
* BFN modelliert den Prozess von Verteilung zu Verteilung, die Eingabe ist die Profildarstellung und die Ausgabe ist immer noch die Profildarstellung
* Anstatt von Grund auf neu zu argumentieren, kann BFN Profilinformationen als a priori für bedingtes Denken einführen
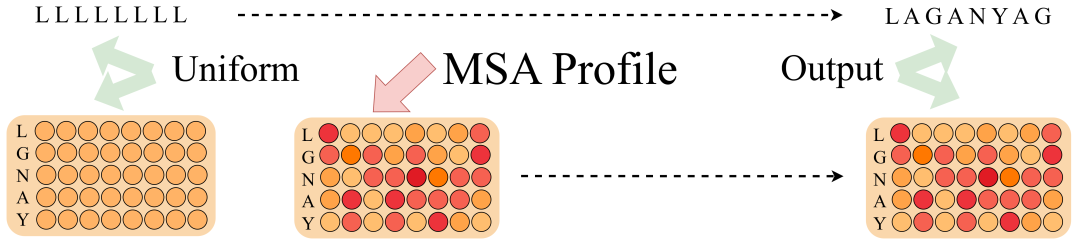
Herkömmliche Modelle wie das autoregressive Modell und das Diffusionsmodell erfordern Daten (Token) als Eingabe, und die Verarbeitung von Profilinformationen erhöht die Komplexität des Algorithmus.
Mit BFN als Modellskelett kann ProfileBFN außerdem Folgendes erreichen:
* Vereinfachung von Aufgaben. Die bedingte Generierung homologer Informationen wird zur Nachahmung von Profilinformationen.
* Verbesserte Effizienz. Der Probenbereich wird reduziert und die Effektivität verbessert
ProfileBFN gilt als Retter der Nassprüfung
Bei Aufgabenstellungen wie der synthetischen Biologie sind lange Zyklen, einzelne Bewertungsindikatoren und mangelnde Glaubwürdigkeit häufige Probleme, mit denen Forscher konfrontiert werden. Als Proteinbasismodell kann ProfileBFN mehr homologe Informationen bei begrenzten Ressourcen integrieren, spezifische Vorinformationen voll ausnutzen und hat einen guten Migrationseffekt auf mehrere Indikatoren, was es zweifellos zur besten Wahl für die Synthese von Kandidatenproteinen und die gerichtete Evolution macht.
Über die Forschungsgruppe
Das Forschungsfeld der Generative Symbolic Intelligence Research Group (GenSI) des Tsinghua University Institute of Intelligent Industry umfasst die beiden Richtungen LLM und AI for Science. Es wird erwartet, dass sich die beiden Richtungen gegenseitig fördern und so die ultimative Mission der AGI für die Wissenschaft (AI Scientist) erreichen.
Zu den spezifischen Forschungsrichtungen gehören die neue Generation groß angelegter Vortrainingstechnologie, groß angelegtes bestärkendes Lernen (Large Scale RL), tiefe generative Modelle (Deep Generative Models) und deren Anwendung in wissenschaftlichen Daten, wobei der Schwerpunkt auf der Integration und Innovation grundlegender Algorithmen der künstlichen Intelligenz und wissenschaftlicher Probleme liegt. Derzeit konzentriert sich das Team auf die neuesten Theorien tiefer generativer Modelle und die Erforschung von Methoden skalierbarer, strukturbasierter generativer Modelle und widmet sich der Lösung realistischer und anspruchsvoller wissenschaftlicher Probleme in den Bereichen LLM und AI4Sci, wie etwa der Verbesserung der Argumentationsfähigkeit von LLM und der Überwindung von Strukturgenerierungsaufgaben auf AF3-Niveau.
Das Team kann über die folgenden Kanäle kontaktiert werden ⬇️
* Homepage:https://go.hyper.ai/7ye91
* E-Mail: [email protected]
* Xiaohongshu/Zhihu: GenSI
* Twitter: @GenSI_official
* WeChat: 15805171115








