Command Palette
Search for a command to run...
Vorhersage Enzymkinetischer Parameter, Identifizierung Von Engpässen ... Luo Xiaozhou Vom Shenzhen Institute of Advanced Technology Der Chinesischen Akademie Der Wissenschaften Berichtete Über Die Innovative Anwendung Von KI Im Enzymbereich

Protein spielt als Grundstein des Lebens eine Schlüsselrolle in allen Lebensaktivitäten. Die Untersuchung seiner Struktur und Funktion ist für die innovative Arzneimittelentwicklung, die synthetische Biologie, die Enzymproduktion und andere Bereiche von großer Bedeutung. Allerdings ist das traditionelle Proteindesign mit zahlreichen Herausforderungen verbunden. Die Proteinstruktur ist komplex und der Sequenzraum ist riesig. Die Designmethode, die auf Expertenerfahrung und Hochdurchsatz-Screening beruht, ist nicht nur zeit- und arbeitsintensiv, sondern garantiert auch kaum eine Erfolgsquote.
Heute ist „KI für die Wissenschaft“ eine neue Grenze in der Entwicklung der globalen künstlichen Intelligenz, die das Paradigma der wissenschaftlichen Forschung grundlegend verändert und enorme Veränderungen im Bereich des Proteindesigns mit sich bringt. Insbesondere nach dem Aufkommen innovativer Ergebnisse wie AlphaFold ist die damit verbundene Forschung allmählich ins öffentliche Bewusstsein gerückt und hat mehr Aufmerksamkeit erhalten. Gleichzeitig wurden im In- und Ausland weitere herausragende Teams gefördert, die sich diesem Thema widmen und Schwierigkeiten aus verschiedenen Aspekten wie Technologie und Anwendung angehen.
Einer von ihnen ist Professor Luo Xiaozhou, Forscher am Shenzhen Institute of Advanced Technology der Chinesischen Akademie der Wissenschaften. Zuvor konzentrierte er sich auf synthetische Biologie. Nach seiner Rückkehr nach China im Jahr 2019 begann er, sich der KI-Proteinforschung zu widmen. Auf dem „Future is Here“-AI-Protein-Design-Gipfel, der kürzlich von der Shanghai Jiao Tong University in China veranstaltet wurde, teilte Professor Luo Xiaozhou seine Ansichten zum Thema „Enzym-Engineering auf Basis künstlicher Intelligenz“. Erkunden Sie die potenziellen Anwendungen von multimodalem Lernen und generativer KI im Enzymdesign,Die innovativen Anwendungen und Praktiken der KI im Bereich der Enzymtechnik werden aus mehreren Perspektiven erläutert, beispielsweise anhand des UniKP-Frameworks und der ProEnsemble-Maschine.

HyperAI hat den ausführlichen Austausch organisiert und zusammengefasst, ohne die ursprüngliche Absicht zu verletzen. Nachfolgend finden Sie eine Abschrift der wichtigsten Punkte der Rede.
Automatisierter Plattformaufbau, KI löst Proteinprobleme
Naturprodukte sind eine wahre Fundgrube medizinischer Materialien und zeichnen sich durch breite Quellen, reichhaltige Strukturen und vielfältige Wirkungen aus. Allerdings ist die traditionelle Methode zur Extraktion von Naturprodukten aus natürlichen Ressourcen ineffizient und die reine chemische Synthese liefert nicht nur geringe Erträge, sondern erfordert auch den Einsatz großer Mengen giftiger und gefährlicher Reagenzien. Beispielsweise wurde Artemisinin ursprünglich aus Artemisia annua extrahiert, bei der chemischen Synthese traten jedoch viele Probleme auf. Später wurde die Artemisininexpression in Saccharomyces cerevisiae durch die Regulierung mehrerer Gene erreicht. Dieser Durchbruch ermöglichte es uns, das Potenzial der Biosynthese zu erkennen, und so begann ich, mich auf die Forschung im biologischen Bereich zu konzentrieren. Darüber hinaus wird der Forschungsfortschritt im Bereich der Enzymmodifikation durch den Mangel an Daten erheblich eingeschränkt. Dieses Problem macht uns die Bedeutung von Daten bewusst. Daher setze ich mich für den Aufbau von Automatisierungs- und Datenplattformen ein, um den Grundstein für die spätere KI-Forschung zu legen.
Als Grundmoleküle des Lebens werden Nukleinsäuren, niedermolekulare Lipide, Kohlenhydrate, Metabolite, Ionen, Wasser und andere Substanzen aus Proteinen hergestellt. Aufgrund dieser Eigenschaft habe ich nach meiner Rückkehr nach China im Jahr 2019 meine Forschung auf den Bereich der Proteine konzentriert und drei wissenschaftliche Fragen aufgeworfen: Erstens: Ist es möglich, die Aktivität und Funktion eines Proteins direkt anhand seiner Sequenz vorherzusagen? Die zweite Frage ist, ob es möglich ist, die von Menschen benötigten Proteine bei Bedarf zu erzeugen oder weiterzuentwickeln. Die dritte Frage ist, ob es möglich ist, Enzyme oder Stämme auf der Grundlage einer universellen, standardisierten Strategie zu optimieren?
UniKP-Framework sagt Enzymeigenschaften besser voraus
Im Lehrbuch heißt es: Die Primärsequenz eines Proteins bestimmt seine Tertiärstruktur und Funktion, und die Primärsequenz muss funktionelle Informationen enthalten. Daher ist die Art und Weise, wie die Sequenz extrahiert wird, äußerst wichtig. Inspiriert von AlphaFold begann unser Team, Methoden zu erforschen, um die Funktion von Proteinen anhand von Sequenzen vorherzusagen. In unserer Studie haben wir die Transformer-Architektur eingeführt, um traditionelle Darstellungsmethoden mit Funktionen des maschinellen Lernens zu integrieren und so ein integriertes Modell zu erstellen.Das auf Fusionsmerkmalen und integrierten Modellen basierende Framework zur Vorhersage von Peptid- und Proteinfunktionen hat bei 8 verwandten Vorhersageaufgaben eine SOTA-Leistung erreicht und Peptid- und Proteinfunktionen präzise vorhergesagt.Es beschleunigt den Screening-Prozess von Wirkstoffen gegen Infektionen, wie beispielsweise antimikrobiellen Peptiden, und reduziert die Versuchskosten.
Anschließend versuchte das Team mithilfe des UniKP-Frameworks, die Eigenschaften von Enzymen auf Grundlage des Transformer-Embedding-Tools zur Vorhersage enzymatischer Parameter vorherzusagen. Verwenden Sie ProtT5 und das traditionelle SMILE-Transformer-Modell, um die Sequenz zu vektorisieren und sie mit einem einfachen maschinellen Lernmodell zu kombinieren, um SOTA-Ergebnisse zu erzielen.
Das Forschungsteam wählte vier repräsentative Datensätze aus, um die Leistung und den Wert von UniKP zu überprüfen.
Zuerst ist der DLkcat-Datensatz,Die Forscher untersuchten 16.838 Proben, darunter 7.822 einzigartige Proteinsequenzen und 2.672 einzigartige Substrate von 851 Organismen. Der Datensatz wird im Verhältnis 9:1 in Trainingsdatensatz und Testdatensatz aufgeteilt.
Als nächstes folgen die pH- und Temperaturdatensätze.Der pH-Datensatz enthält 636 Proben, bestehend aus 261 einzigartigen Enzymsequenzen und 331 einzigartigen Substraten; Der Temperaturdatensatz enthält 572 Proben, bestehend aus 243 einzigartigen Enzymsequenzen und 302 einzigartigen Substraten. Der Datensatz wird im Verhältnis 8:2 in Trainingsdatensatz und Testdatensatz aufgeteilt.
Der dritte ist der Datensatz der Michaelis-Konstante (Km),Es besteht aus 11.722 Proben, einschließlich Enzymsequenzen, molekularen Fingerabdrücken des Substrats und entsprechenden Km-Werten. Der Datensatz wird im Verhältnis 8:2 in Trainingsdatensatz und Testdatensatz aufgeteilt.
Der vierte ist der kcat/Km-Datensatz,Enthält 910 Proben, bestehend aus Enzymsequenzen, Substratstrukturen und ihren entsprechenden kcat/Km-Werten.
Es wurde nachgewiesen, dass UniKP bei der kcat-Vorhersage deutlich besser ist als bestehende Modelle und erstmals eine kcat/Km-Vorhersage erreicht.Am Beispiel von kcat liegt der Determinationskoeffizient im größten öffentlich verfügbaren Datensatz um 20 Prozentpunkte höher als das aktuelle SOTA-Ergebnis. Gleichzeitig ist die Leistung auch bei mehreren Aufgaben, wie beispielsweise bei unterschiedlichen Datensatzunterteilungen, unterschiedlichen Intervallunterteilungen und unterschiedlichen Enzymkategorieunterteilungen, deutlich besser.
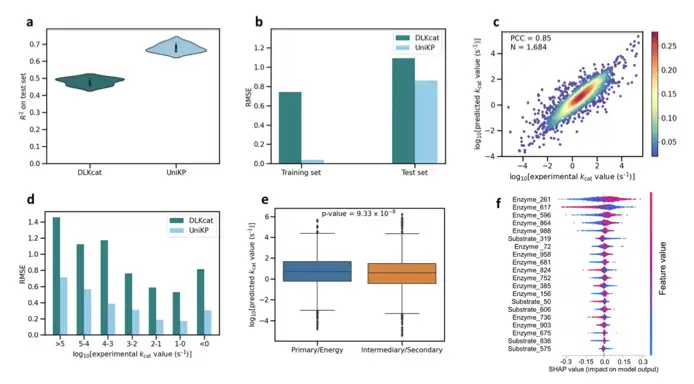
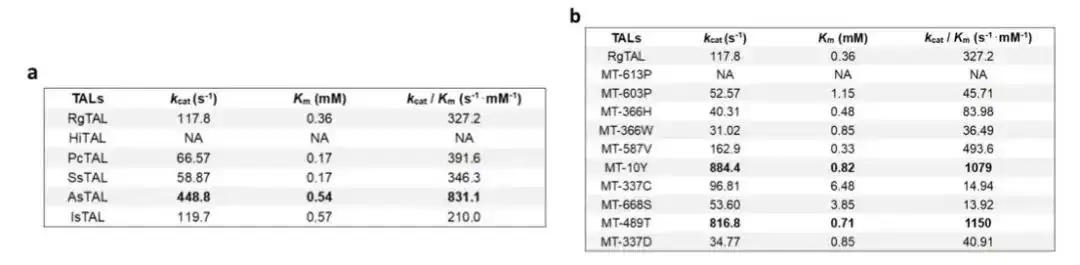
Mithilfe dieser Architektur haben wir aus 1.000 Blast-Sequenzen das Wildtyp-TAL-Enzym mit der bislang höchsten Enzymaktivität gefunden und durch die Vorhersage von Einzelstellenmutationen Mutanten mit höherer Enzymaktivität erhalten, wodurch der Prozess der Enzymentwicklung erheblich beschleunigt wurde.
Darüber hinaus haben wir mit Blick auf die thermische Stabilität von Proteinen ein sequenzbasiertes Vorhersagemodell für thermophile Proteine namens Thermal Finer vorgeschlagen, das bei drei Klassifizierungsdatensätzen eine SOTA-Leistung erreichte und erstmals die Vorhersage der entsprechenden optimalen katalytischen Temperatur (Regression) basierend auf der Proteinsequenz ermöglichte. Mit anderen Worten: Zum ersten Mal ist es uns gelungen, die optimale Temperatur direkt aus der Proteinsequenz vorherzusagen, was die Gewinnung und Entwicklung von Enzymen stark unterstützt.
Feinabstimmung von ProGPT-2 zur Erzeugung oder Entwicklung von Proteinen nach Bedarf
Derzeit gibt es zwei Haupttypen von Modellen für die Proteinproduktion, insbesondere die Enzymproduktion:
* Generative Adversarial Neural Networks (GAN): ProteinGAN
* Vortrainierte generative große Sprachmodelle (LLM): ProtGPT2, ProGen
Aber,Bei allen diesen Werkzeugen zur Proteinerzeugung besteht das Problem, dass sie ähnliche Sequenzen erzeugen und den Anforderungen an die Erzeugung von Enzymen mit neuen Funktionen und neuen Aktivitäten nicht gerecht werden.Es gibt auch einige unvernünftige Aspekte in der theoretischen Analyse: Erstens sind die Pixelwerte des Bildes kontinuierlich, was für die Gradientenoptimierung besser geeignet ist; zweitens ist der Text (Aminosäuresequenz) diskontinuierlich und die Gradientenoptimierung ist für die Aktualisierung von Einbettungen bedeutungslos und sehr ineffizient.
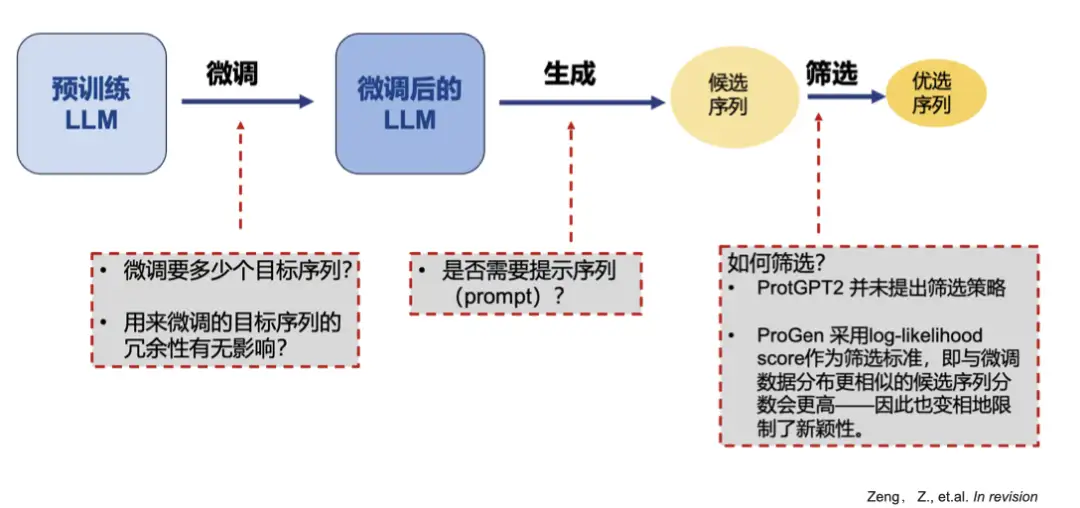
Für solche Probleme analysieren wir gründlich die Mängel bestehender Modelle und schlagen ein neues Optimierungsframework vor.
Unser Team hat ProGPT-2 feinabgestimmt und ein neuronales CNN-Netzwerk als Diskriminator verwendet, um die generierten Sequenzen zu filtern und zu priorisieren. Durch Experimente wurde festgestellt, dassZur Feinabstimmung der Sequenz sind nur 2000 oder sogar weniger erforderlich, und die generierte Sequenz ohne Hinweiswörter kommt dem natürlichen Enzym näher. Gleichzeitig kann durch die Reduzierung redundanter Daten der Neuartigkeitsgrad der generierten Sequenzen verbessert werden.
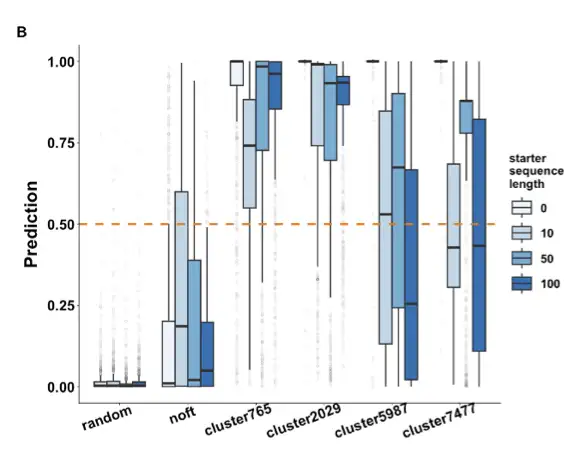
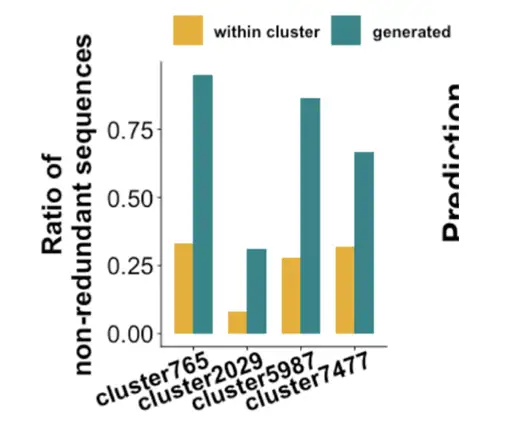
Wir wollen neuartige Enzyme mit neuen Strukturen und Funktionen, daher brauchen wir keine redundanten Sequenzen. Durch prädiktive antimikrobielle Peptide stellten wir fest, dass das Modell grundsätzlich gut funktionierte. Anschließend führten wir eine MDH-Analyse durch und stellten Folgendes fest:Die wichtigsten Stätten sind stark konserviert; die Vorhersagewerte derjenigen mit Signaturdomänen sind höher; und die Ergebnisse des molekularen Dockings sind grundsätzlich dieselben wie die von MDH in der Natur.Wie in der folgenden Abbildung dargestellt:
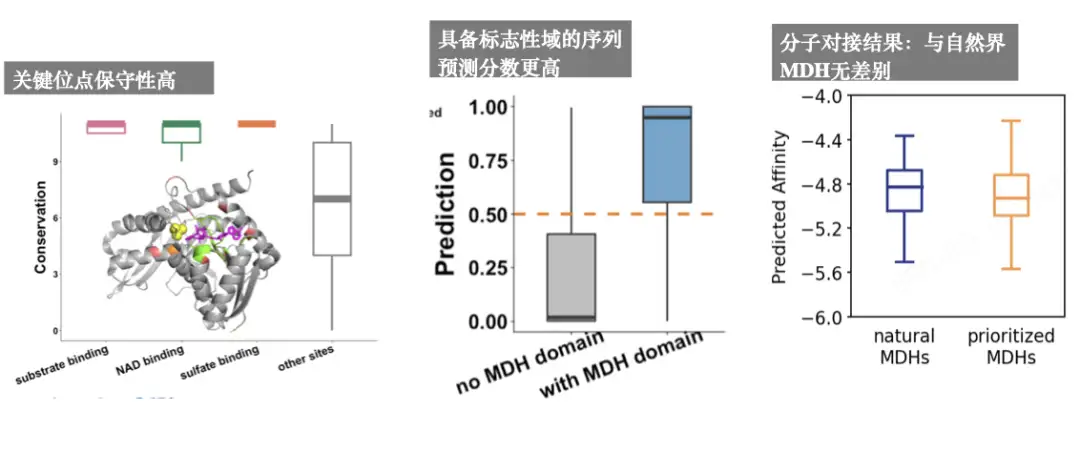
Anschließend überprüften wir, ob die nach dem Modell produzierten ungleichen Enzyme funktionsfähig waren. Basierend auf den Originaldaten von ProteinGAN können die Enzyme mit einer Ähnlichkeit von 80% nach dem priorisierten MDHs-Modell eine Ähnlichkeit von weniger als 40% erreichen. Im Vergleich zu den 10 Enzymen, die wir zufällig aus der Natur ausgewählt haben, ist es hinsichtlich Unlöslichkeit, Nichtexpression und Löslichkeit grundsätzlich dasselbe, weist aber dennoch eine sehr gute Enzymaktivität auf. Mit anderen Worten:Die von unserem Team mithilfe dieses Modells erzeugten Enzyme weisen eine geringe Ähnlichkeit mit natürlichen Enzymen auf und die meisten von ihnen weisen eine enzymatische Aktivität auf.
ProEnsemble Stoffwechselengpässe erkennen und Enzymproduktion optimieren
Im Biosyntheseprozess machen eine Reihe metabolischer Engpässe, wie etwa die geringe katalytische Effizienz mehrerer Enzyme im Stoffwechselweg und epistatische Effekte zwischen Enzymen, den Optimierungsprozess komplex und unsicher. Eine Überexpression von Stoffwechselwegenzymen beeinträchtigt häufig das Zellwachstum und die Produktexpression, und einige Enzyme können negative Auswirkungen haben. Zu diesem Zweck habe ich gefragt, ob es eine universelle, standardisierte Strategie zur Optimierung von Enzymen oder Stämmen gibt?
Lassen Sie uns zunächst überprüfen, ob eine Überexpression wirklich schlecht ist.Das Team reduzierte künstlich die Expressionsniveaus bestimmter Enzyme, um künstliche Stoffwechselengpässe zu erzeugen und so kontrollierbaren evolutionären Raum zu erhalten.
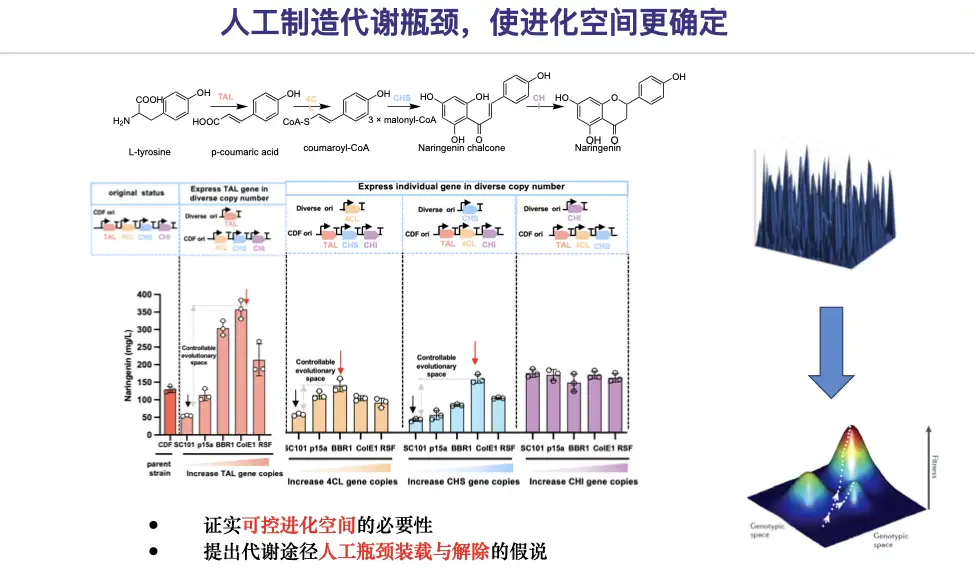
Daher wird am Beispiel von Naringenin eine Lösung für die Gestaltung von Signalweg-Engpässen und eine Strategie zur Eliminierung vorgeschlagen:
* In der ersten Phase werden wir die automatisierte Plattformtechnologie für Großanlagen nutzen, um die Expression der mit der Naringeninsynthese verbundenen Gene auf niedrigem Niveau (niedriger Kopienzahlhintergrund) zu ermöglichen und so einen künstlichen metabolischen Engpass für die Naringeninsynthese zu konstruieren.
* Im zweiten Schritt wurden die Kandidatenmutanten 4CL-11C1 und CHS-9H9 auf ihre Naringeninproduktion untersucht, die mit der der ursprünglichen Mutanten vergleichbar war, wodurch der Engpass des Naringenin-Stoffwechselwegs beseitigt wurde.
* Im dritten Schritt werden durch KI-vermitteltes Promotor-Engineering Mutanten einzelner Gene wieder in den ursprünglichen Stoffwechselweg zurückgeführt und der Stoffwechselfluss ausgeglichen.
Die Ergebnisse der Studie zeigten, dassStrategien zur künstlichen Schaffung und Beseitigung von Engpässen ermöglichen eine effiziente Entwicklung von Stoffwechselwegen innerhalb klarer Bahnen.Darüber hinaus wird bestätigt, dass epistatische Effekte die Grenzen der Pfadentwicklung begrenzen können.
Auf dieser Grundlage haben wir einen automatisierten Prozess entwickelt, der Anweisungen, Klonen und bakterielle Screeningtests umfasst.Die Ergebnisse zeigten, dass es hinsichtlich Wachstum, Siebung und Produktgewinnung keinen signifikanten Unterschied zwischen diesem Verfahren und der manuellen Vorgehensweise gab.Allerdings sind Methoden zur Automatisierung der Stoffwechselweg-EvolutionDie Zeit für die parallele Evolution mehrerer Enzyme wird erheblich verkürzt und eine Runde der parallelen Evolution kann innerhalb von zwei Wochen abgeschlossen werden.
Auf der Grundlage einer großen Datenmenge entwickelte das Team ein integriertes maschinelles Lernmodell namens ProEnsemble zur Optimierung metabolischer Zuwächse. Experimente zeigten, dass das integrierte Modell auf Basis maschinellen Lernens die Stoffwechselwege ausgleicht und die Naringeninproduktion im Vergleich zum nicht optimierten Modell um das 5,16-fache steigert. Es erreichte 1,21 g/l in einer 96-Well-Platte und 3,65 g/l in einem Fermenter und damit den höchsten gemeldeten Wert. Allein durch die Überexpression wichtiger synthetischer Gene war die Produktion verschiedener modifizierter zusammengesetzter Chassis höher als die in der Literatur berichteten Werte (mithilfe von Strategien des Stoffwechsel-Engineerings).
Die Lernstrategie von ProEnsemble baut ein geschlossenes Kreislaufsystem zur Identifizierung und Optimierung metabolischer Engpässe auf und entwickelt erfolgreich ein Naringenin-Chassis mit hoher Ausbeute für Escherichia coli, das um ein Vielfaches höher ist als das aktuelle Niveau in der Branche und eine universelle Lösung für das Gleichgewicht komplexer metabolischer Netzwerke bietet.
Aufbau einer groß angelegten Automatisierungsplattform zur Förderung der Zusammenarbeit zwischen Industrie, Universitäten und Forschung
Abschließend möchte ich Ihnen die industrielle Umsetzung dieser Errungenschaften vorstellen. Wir haben eine groß angelegte, vollautomatische Plattform aufgebaut – die große wissenschaftliche und technologische Einrichtung für synthetische Biologieforschung in Shenzhen, China, die eine groß angelegte automatisierte Plattform umfasst, die mehrere Plattformen wie Designlernen, synthetische Tests und Benutzertests abdeckt. Die Plattform verfügt über leistungsstarke Funktionen und kann standardisierte Datenverarbeitung und experimentelles Design für maschinelles Lernen in der Cloud durchführen. Roboter können bei der Durchführung experimenteller Vorgänge behilflich sein. Die Spektrumvorbereitung und die Erkennungsgeschwindigkeit sind hoch und eine Probe kann in nur 10 Sekunden generiert werden, wodurch eine Hochdurchsatzerkennung erreicht wird.
Darüber hinaus bietet die Plattform auch automatisiertes, unterstütztes Softwaredesign, sodass Benutzer die erforderlichen Komponenten direkt aus der Komponentenbibliothek auswählen und experimentelle Anweisungen generieren können. Wir haben mittlerweile mit vielen Industrie- und Hochschuleinrichtungen zusammengearbeitet. Wir sind die erste Plattform in der Branche, die den vollständigen Prozess der Streptomyces-Automatisierung realisiert. Wir heißen jeden willkommen, mit uns zusammenzuarbeiten.
Über Professor Luo Xiaozhou
Professor Luo Xiaozhou ist Forscher, Doktorvater und stellvertretender Direktor des Instituts für Synthetische Biologie am Shenzhen Institutes of Advanced Technology der Chinesischen Akademie der Wissenschaften. Er ist ein ausgewählter Experte des National Major Talent Project – Youth Project, CTO des National Biomanufacturing Industry Innovation Center und stellvertretender Chefprozessingenieur der Shenzhen Major Science and Technology Facility for Synthetic Biology, China.
Er erhielt seinen Ph.D. in Chemie vom Scripps Research Institute im Jahr 2016 (Betreuer: Akademiker Peter G. Schultz) und absolvierte anschließend eine Postdoc-Forschung an der University of California, Berkeley (Co-Betreuer: Akademiker Jay D Keasling). Im Jahr 2019 wechselte er zum Shenzhen Institutes of Advanced Technology der Chinesischen Akademie der Wissenschaften. Er wurde für das National Youth Talent Program, die Guangdong Province Outstanding Young Scholars und die Shenzhen City Excellent Young Scholars ausgewählt.
Seine Forschung konzentriert sich auf die biochemischen Prozesse in lebenden Organismen im Bereich der synthetischen Biologie, einschließlich der gerichteten Evolution von Enzymen, Protein-Engineering, Hochdurchsatz-Screening und der Totalbiosynthese natürlicher und nicht-natürlicher Verbindungen. Als korrespondierender Autor hat er 20 Artikel in Nature Metabolism, Advanced Science, Nature Synthesis, Nature Communications und Angew. veröffentlicht. Chem. Int. Ed. usw. und insgesamt mehr als 50 SCI-Papiere, mehr als 30 Patente angemeldet und 6 genehmigt.