Command Palette
Search for a command to run...
Das Team Der Westlake University Hat SaProt Und Andere Proteinsprachenmodelle Als Open Source Bereitgestellt Und Deckt Damit Die Vorhersage Von Strukturfunktionen, Die Modalübergreifende Informationssuche, Das Design Von Aminosäuresequenzen usw. ab.

Am 22. und 23. März 2025 fand offiziell der „AI Protein Design Summit“ der Shanghai Jiao Tong University statt.Der Gipfel brachte mehr als 300 Experten und Wissenschaftler von namhaften Universitäten wie der Tsinghua-Universität, der Peking-Universität, der Fudan-Universität, der Zhejiang-Universität und der Xiamen-Universität sowie mehr als 200 Vertreter führender Industrieunternehmen und technisches F&E-Personal zusammen, um die neuesten Forschungsergebnisse, technologischen Durchbrüche und industriellen Anwendungsaussichten von KI im Bereich des Proteindesigns eingehend zu diskutieren.

Während des GipfelsDr. Yuan Fajie von der Westlake University berichtete unter dem Thema „Forschung und Anwendung von Proteinsprachmodellen“ über die neuesten Forschungsfortschritte bei Proteinsprachmodellen und stellte die wichtigen Errungenschaften des Teams im Detail vor.Einschließlich der Proteinsprachenmodelle SaProt, ProTrek, Pinal, Evolla usw. HyperAI hat den ausführlichen Austausch organisiert und zusammengefasst, ohne die ursprüngliche Absicht zu verletzen. Nachfolgend finden Sie eine Abschrift der wichtigsten Punkte der Rede.
Ein bemerkenswertes Proteinsprachenmodell
Proteine sind biologische Makromoleküle, die aus 20 in Reihe geschalteten Aminosäuren bestehen. Sie erfüllen im Körper wichtige Funktionen wie Katalyse und Stoffwechsel und sind die Hauptausführer der Lebensaktivitäten. Biologen unterteilen die Struktur von Proteinen üblicherweise in vier Ebenen: Die Primärstruktur beschreibt die Aminosäuresequenz des Proteins, die Sekundärstruktur konzentriert sich auf die lokale Konformation des Proteins, die Tertiärstruktur stellt die gesamte dreidimensionale Konfiguration des Proteins dar und die Quartärstruktur beinhaltet die Interaktion zwischen mehreren Proteinmolekülen.Im Bereich der AI-Proteine konzentriert sich die Forschung vor allem auf diese Strukturen.
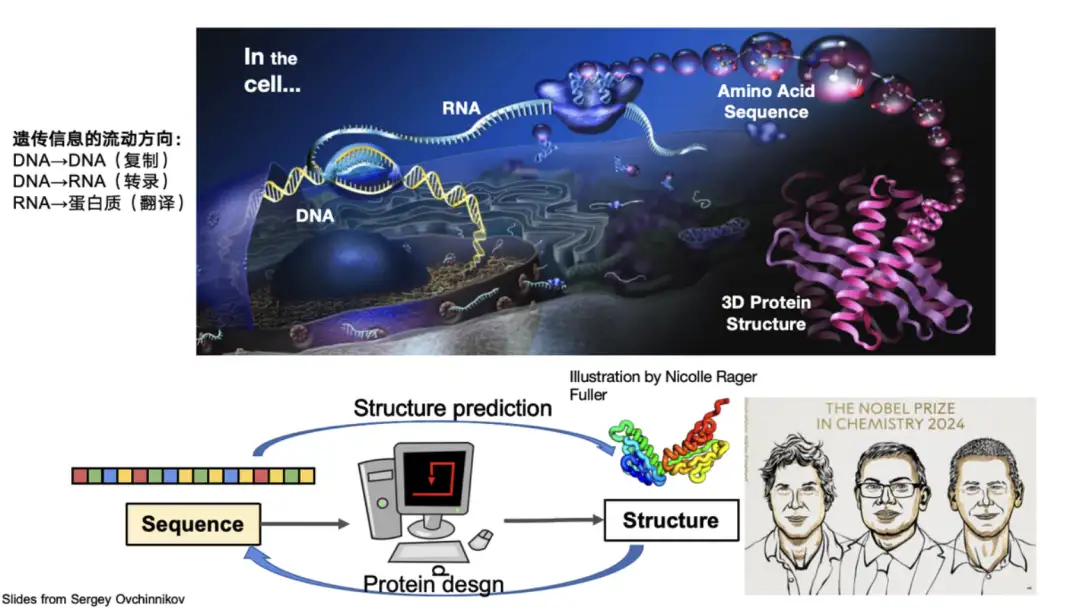
Beispielsweise ist die Vorhersage der dreidimensionalen Struktur eines Proteins anhand seiner Sequenz das Kernproblem, das AlphaFold 2 löst. Es hat das Proteinfaltungsproblem überwunden, das die wissenschaftliche Gemeinschaft 50 Jahre lang geplagt hat, und wurde dafür mit dem Nobelpreis ausgezeichnet. Andererseits erhielt auch Professor David Baker, ein wichtiger Mitarbeiter auf dem Gebiet des Proteindesigns, also der Entwicklung neuer Proteinsequenzen auf der Grundlage von Struktur und Funktion, den Nobelpreis.
Traditionell werden Proteinstrukturen üblicherweise in Form von PDB-Koordinaten dargestellt. In den letzten Jahren haben Forscher Methoden erforscht, um kontinuierliche räumliche Strukturinformationen in diskrete Token umzuwandeln, wie etwa Foldseek, ProTokens, FoldToken, ProtSSN, ESM-3 usw.
*Foldseek kann die dreidimensionale Struktur eines Proteins in eindimensionale diskrete Token kodieren.
Das Proteinsprachenmodell unseres Teams basiert auf diesen diskreten Ergebnissen.
Der Großteil der Forschung zu KI und Proteinen lässt sich auf die Forschung zur natürlichen Sprachverarbeitung zurückführen. Lassen Sie uns daher zunächst zwei klassische Sprachmodelle im Bereich der natürlichen Sprachverarbeitung (NLP) betrachten:Eines ist das unidirektionale Sprachmodell, das durch die GPT-Reihe dargestellt wird.Sein Mechanismus basiert auf dem Informationsfluss von links nach rechts und sagt das nächste Token anhand der Daten auf der linken Seite (oben) voraus.Eines ist das bidirektionale Sprachmodell, das durch BERT dargestellt wird.Es ist durch das Masked Language Model vortrainiert, das die Informationen (den Kontext) auf der linken und rechten Seite eines gekochten Wortes erkennen und das gekochte Wort vorhersagen kann.
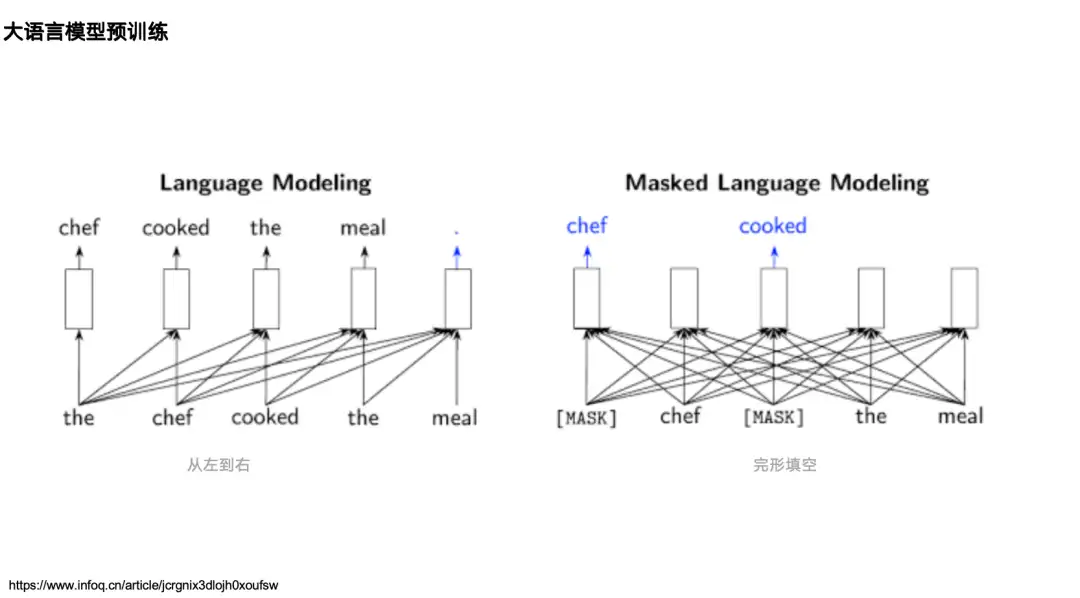
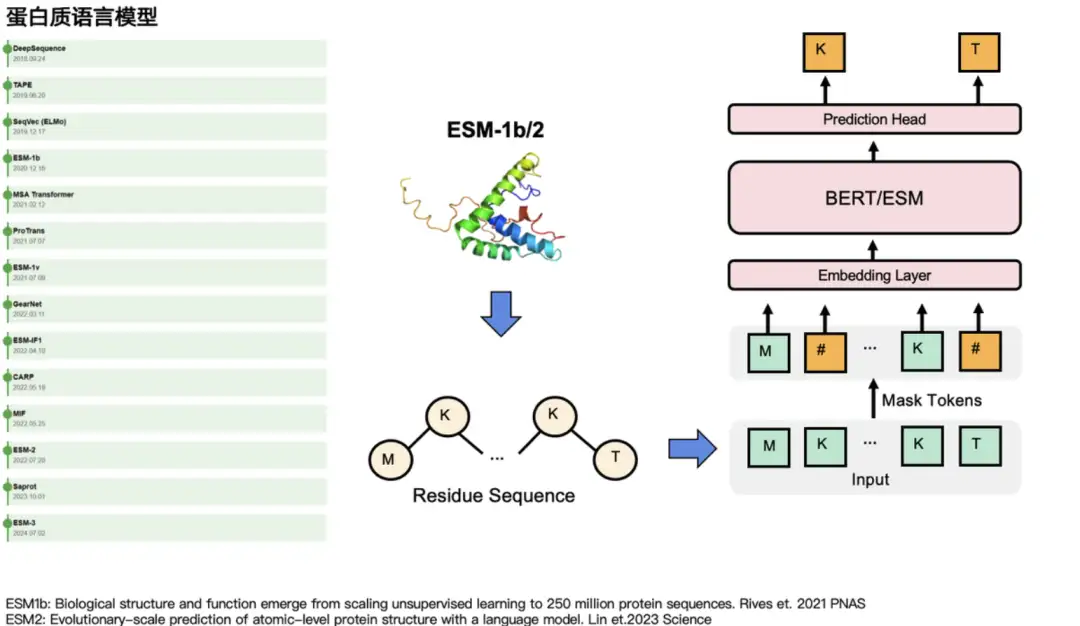
Im Proteinbereich gibt es für beide Modelltypen entsprechende Proteinsprachenmodelle.Beispielsweise gibt es entsprechend GPT ProtGPT2, ProGen usw. Entsprechend BERT gibt es die Modelle der ESM-Reihe: ESM-1b, ESM-2 und ESM-3. Sie maskieren hauptsächlich einige Aminosäuren und sagen deren „wahre Identität“ voraus. Bei Aufgaben zur natürlichen Sprache maskieren sie einige Wörter und sagen sie dann voraus. Wie auf der linken Seite der Abbildung unten gezeigt, gibt es andere Sprachmodelle, die einen relativ großen Einfluss in der Protein-Community haben, darunter MSA Transformer, GearNet, ProTrans usw.
Das für ICLR 2024 ausgewählte Proteinsprachenmodell SaProt integriert strukturelles Wissen
Das erste Ergebnis, das ich Ihnen vorstellen möchte, ist SaProt, ein Proteinsprachenmodell mit strukturbewusstem Vokabular.Dieser Artikel mit dem Titel „SaProt: Protein Language Modeling with Structure-aware Vocabulary“ wurde für ICLR 2024 ausgewählt.
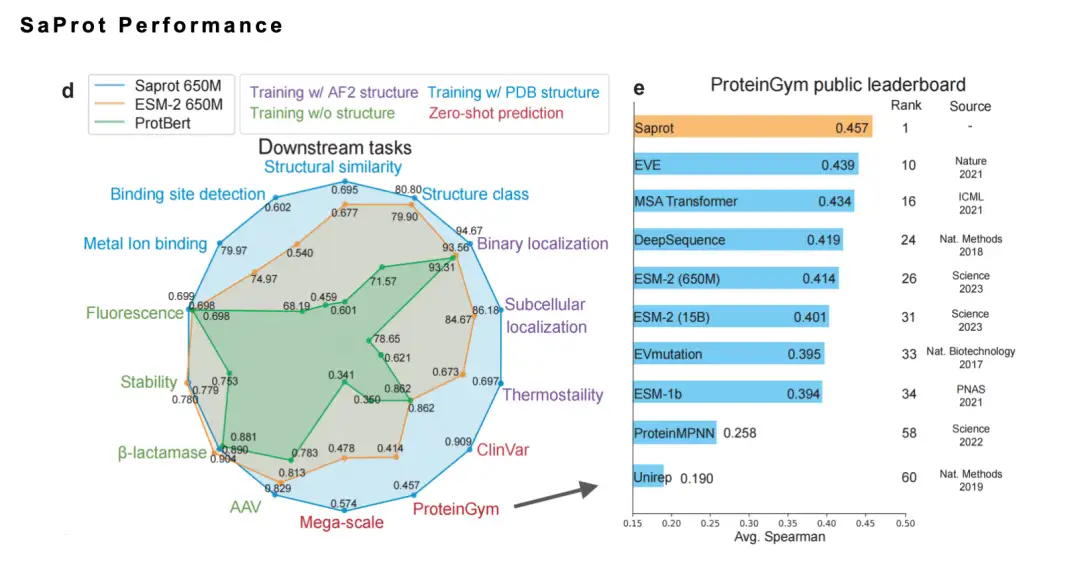
In diesem Artikel haben wir das Konzept eines strukturbewussten Vokabulars vorgeschlagen, Aminosäurerest-Token mit Struktur-Token kombiniert und ein groß angelegtes universelles Proteinsprachenmodell SaProt anhand eines Datensatzes von etwa 40 Millionen Proteinsequenzen und -strukturen trainiert. Dieses Modell übertraf bestehende ausgereifte Basismodelle bei 10 wichtigen nachgelagerten Aufgaben deutlich.
SaProt Open Source-Adresse:
https://github.com/westlake-repl/SaProt
SaProt-Papieradresse:
https://openreview.net/forum?id=6MRm3G4NiU
Warum machen wir dieses Modell?
Tatsächlich basieren die Eingabeinformationen der meisten Proteinsprachenmodelle hauptsächlich auf Aminosäuresequenzen. Nach dem Durchbruch bei AlphaFold arbeitete das DeepMind-Team mit dem Europäischen Bioinformatik-Institut (EMBL-EBI) zusammen, um die AlphaFold-Proteinstrukturdatenbank zu veröffentlichen, in der 200 Millionen Proteinstrukturen gespeichert sind. Also begannen wir zu überlegen: Können wir Proteinstrukturinformationen in das Sprachmodell integrieren, um seine Leistung zu verbessern?

Unser Ansatz ist sehr einfach: Wir verwenden Foldseek, um die Strukturinformationen des Proteins aus der Koordinatenform in diskrete Token umzuwandeln und so ein Aminosäurevokabular und ein Strukturvokabular zu erstellen. Anschließend kombinieren wir diese beiden Vokabulare, um ein neues Vokabular zu generieren, nämlich das strukturbewusste Vokabular (SA-Token). Auf diese Weise kann die ursprüngliche Aminosäuresequenz in eine neue Aminosäuresequenz umgewandelt werden – in dieser Sequenz stellen Großbuchstaben Aminosäure-Token und Kleinbuchstaben Struktur-Token dar. Dann können wir weiter am Masked Language Model arbeiten. Darauf basierend haben wir ein SaProt-Modell mit 650 Millionen Parametern mit 64 A100-GPUs trainiert, die Gesamttrainingszeit betrug etwa 3 Monate.
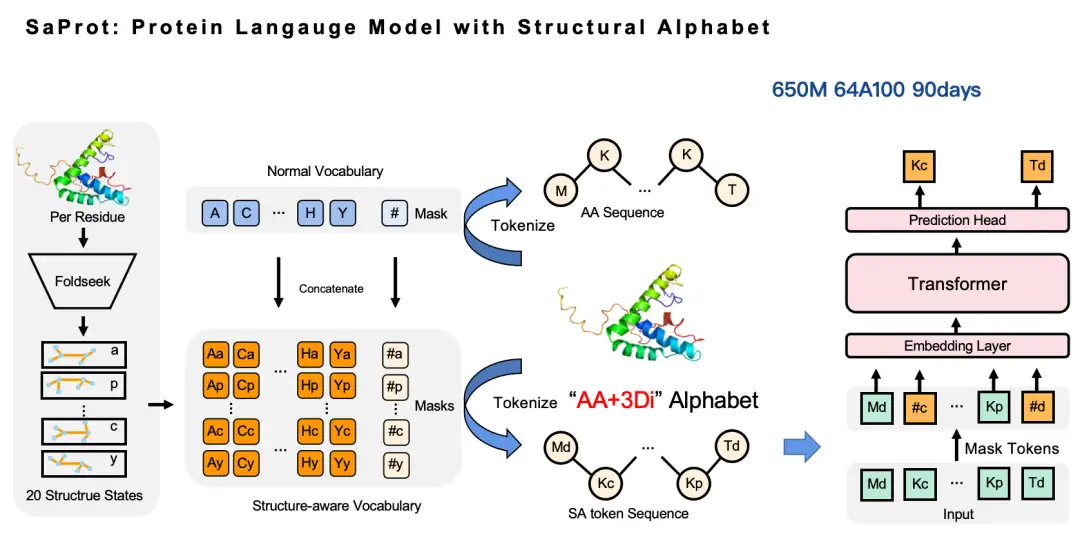
Warum haben wir uns für Foldseek zur Konvertierung von Proteinstruktur-Token entschieden?
Wir haben ein halbes Jahr gebraucht, um uns endlich für die Foldseek 3Di-Token-Sequenz zu entscheiden. Intuitiv sollte die Einbeziehung struktureller Informationen in das Proteinsprachenmodell die Leistung verbessern, aber als wir es tatsächlich versuchten, probierten wir verschiedene Methoden aus, scheiterten jedoch. Beispielsweise haben wir die GNN-Methode zur Modellierung der Proteinstruktur verwendet. Da es sich bei der Proteinstruktur tatsächlich um ein Graph-Neuralnetzwerk handelt, möchten wir die Proteinstruktur natürlich als Graph modellieren und haben daher die MIF-Methode übernommen, jedoch festgestellt, dass das trainierte Modell eine schlechte Generalisierungsfähigkeit aufweist und nicht auf die echte PDB-Struktur erweitert werden kann. Nach eingehender Analyse glauben wir, dass dies möglicherweise daran liegt, dass die Modellierungsmethode unter Verwendung des Masked Language Model zu Problemen mit Informationslecks führt.
Einfach ausgedrückt weist die von AlphaFold vorhergesagte Proteinstruktur selbst bestimmte Verzerrungen, Muster und Spuren von KI-Vorhersagen auf. Wenn diese Daten zum Trainieren eines Sprachmodells verwendet werden, kann das Modell diese Spuren leicht erfassen, was dazu führt, dass das Modell mit den Trainingsdaten zwar eine gute Leistung erbringt, jedoch über eine schlechte Generalisierungsfähigkeit verfügt.
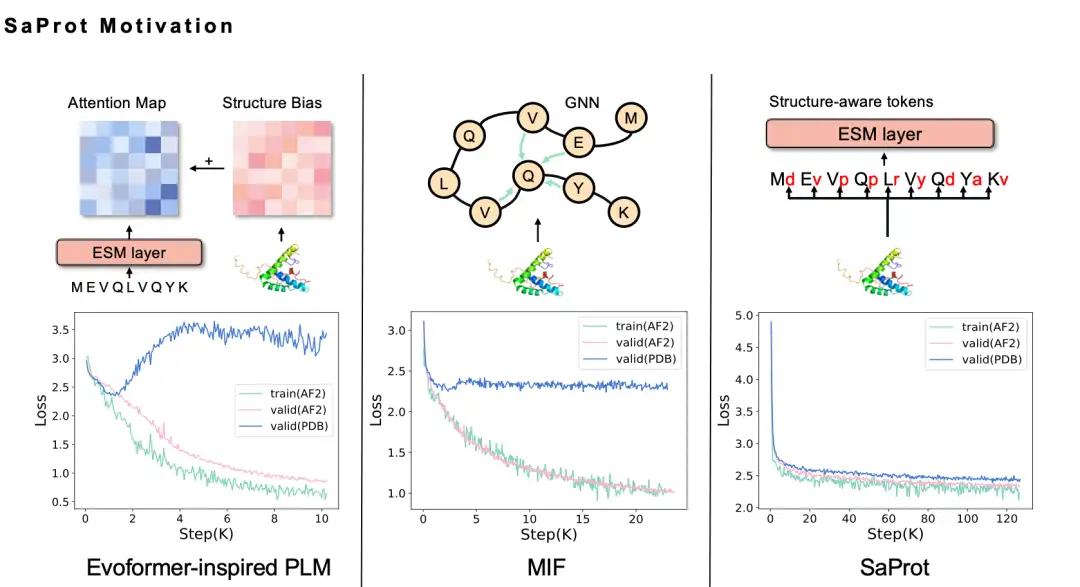
Wir haben verschiedene Verbesserungen ausprobiert, unter anderem die Verwendung der Evoformer-Methode, aber das Problem des Informationsverlusts bestand weiterhin, bis wir Foldseek ausprobierten. Wir haben festgestellt, dass der Verlust des SaProt-Modells, das auf den von AlphaFold vorhergesagten Strukturdaten basiert, reduziert wurde und der Verlust der realen PDB-Strukturdaten ebenfalls deutlich reduziert wurde, was unseren Erwartungen entsprach.
Darüber hinaus schneidet SaProt bei mehreren Benchmarks gut ab.Letztes Jahr belegte es außerdem den ersten Platz auf der maßgeblichen Liste ProteinGym. Gleichzeitig haben wir auch die Ergebnisse der Nassexperiment-Verifizierung von SaProt/ColabSaProt der Community für mehr als 10 Proteine (wie z. B. verschiedene Enzymmutationsmodifikationen, fluoreszierende Proteinmodifikation und Fluoreszenzvorhersage usw.) gesammelt, die alle hervorragende Ergebnisse lieferten.
Obwohl wir das SaProt-Modell für ziemlich gut halten,Aber wenn man bedenkt, dass viele Biologen keine Ausbildung im Deep Learning erhalten haben,Für sie ist es sehr schwierig, ein Proteinsprachenmodell mit etwa einer Milliarde Parametern selbstständig zu optimieren.Deshalb haben wir eine interaktive Schnittstellenplattform namens ColabSaprot + SaprotHub erstellt.
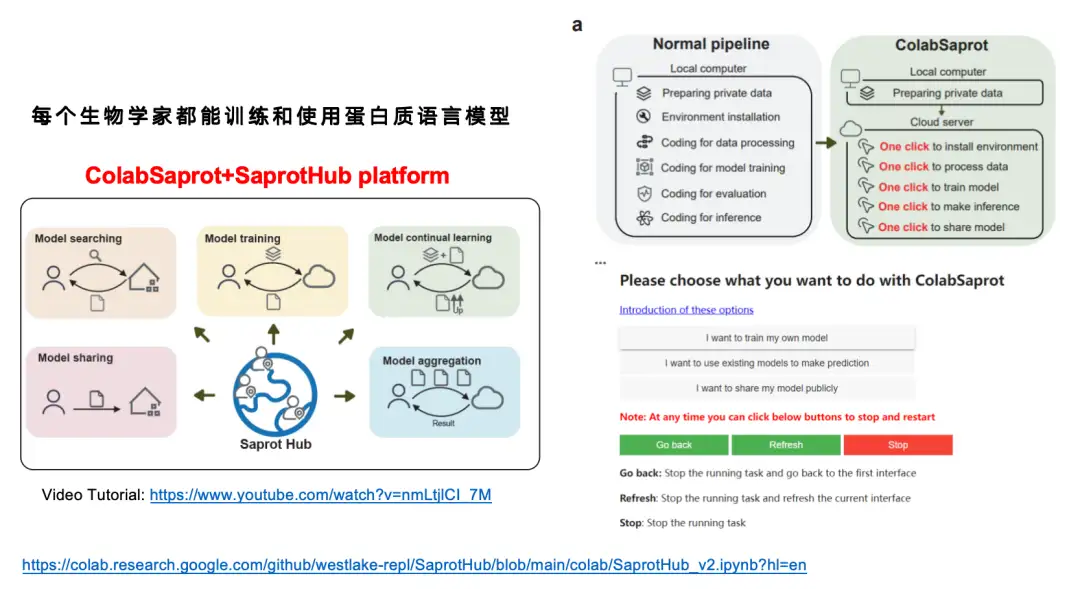
Im herkömmlichen Modelltrainingsprozess (normale Pipeline) müssen Benutzer mehrere Schritte durchlaufen, darunter Datenvorbereitung, Umgebungskonfiguration, Codeschreiben, Datenverarbeitung, Modelltraining, Modellbewertung, Modellbegründung usw. Mit ColabSaprot wurde der gesamte Prozess erheblich vereinfacht – Benutzer müssen nur auf wenige Schaltflächen klicken, um die Umgebungsinstallation, das Modelltraining, die Vorhersage und andere Vorgänge abzuschließen, wodurch die Hemmschwelle für die Verwendung erheblich gesenkt wird.
Wie in der folgenden Abbildung dargestellt, besteht ColabSaprot hauptsächlich aus drei Teilen: Trainingsmodul, Vorhersagemodul und Freigabemodul.

* Im Trainingsmodul müssen Benutzer nur die Aufgabe links beschreiben, die Daten hochladen und dann auf „Trainieren“ klicken. Das System wählt automatisch die optimalen Hyperparameter (wie Batchgröße usw.) aus.
* Im Vorhersagemodul können Benutzer ihre zuvor trainierten Modelle direkt laden und Vorhersagen treffen. Sie können auch von anderen Forschern freigegebene Modelle direkt eingeben, um Vorhersagen zu treffen.
* Das Freigabemodul bietet eine Möglichkeit, den Datenschutz beim Teilen von Ergebnissen zu schützen. Die Daten aus vielen Laboren sind äußerst wertvoll und manche Forscher müssen diese Daten möglicherweise für Folgeforschungen verwenden, möchten aber dennoch bestehende Modelle weitergeben. In ColabSaprot können Benutzer nur das Modell selbst freigeben. Da es sich bei dem Modell im Wesentlichen um eine Blackbox handelt, können andere nicht auf die Originaldaten zugreifen.
Angesichts der Tatsache, dass Sprachmodelle normalerweise sehr groß sind, ist es beim Teilen von Modellen nahezu unmöglich, ein Modell mit einer Milliarde Parametern direkt online zu teilen.Aus diesem Grund haben wir einen ausgereiften Adaptermechanismus übernommen.Benutzer müssen nur eine sehr kleine Anzahl von Parametern teilen – normalerweise nur 1% oder 1/1.000 der Parameter des ursprünglichen Modells. Jeder kann Adapter miteinander teilen und die Adapter anderer Leute laden, um darauf basierend Feineinstellungen oder Vorhersagen vorzunehmen. Wenn die Verbesserung gut ist, können neue Adapter erneut gemeinsam genutzt werden, wodurch ein effizienter Mechanismus zur Zusammenarbeit in der Community entsteht und die Forschungseffizienz erheblich verbessert wird.
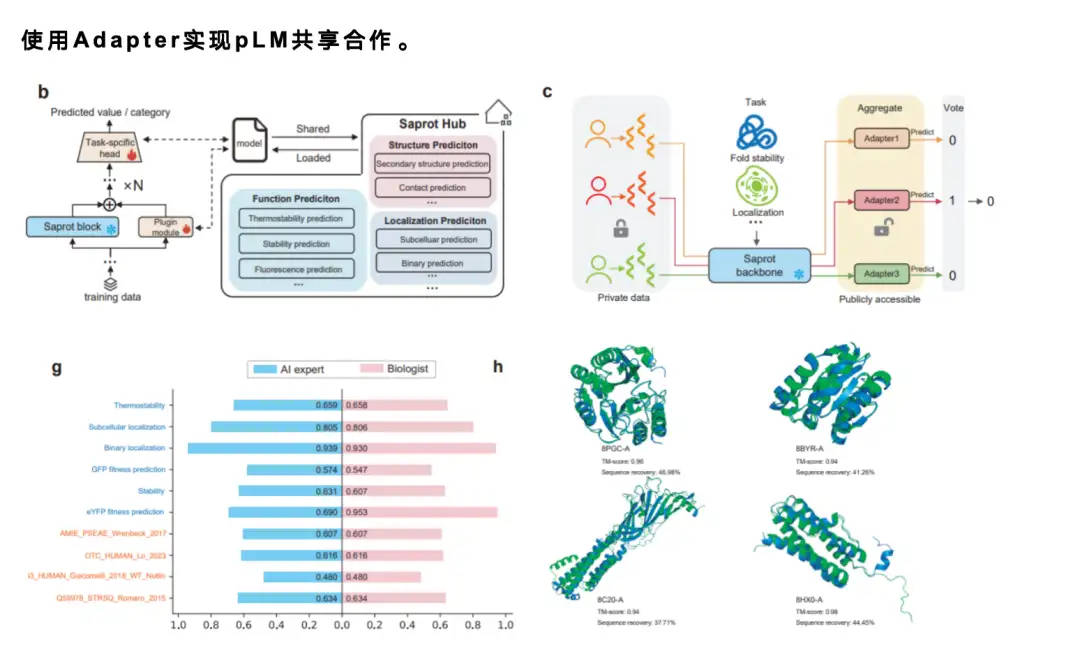
Darüber hinaus haben wir auch eine Benutzerstudie durchgeführt.Wir haben 12 Studenten ohne Vorkenntnisse im maschinellen Lernen oder Programmieren eingeladen, die ColabSaprot-Plattform auszuprobieren. Wir stellten ihnen die Daten zur Verfügung und teilten ihnen die zu erledigenden Aufgaben mit. Sie mussten ColabSaprot für das Modelltraining und die Vorhersage verwenden. Schließlich stellten wir durch den Vergleich ihrer Ergebnisse mit der Leistung von KI-Experten fest, dass diese nicht fachkundigen Benutzer mit ColabSaprot ein Niveau erreichen konnten, das dem von Experten nahe kommt.
Um den Austausch von Proteinsprachenmodellen zu fördern,Wir haben auch eine Community namens OPMC gegründet,Namhafte Wissenschaftler aus dem In- und Ausland auf diesem Gebiet nahmen daran teil und ermutigten alle, Modelle auszutauschen und die Zusammenarbeit und Kommunikation zu fördern.
OPMC-Adresse:

ProTrek-Modell: Finden Sie die Entsprechung zwischen Proteinsequenz, -struktur und -funktion
Die zweite Arbeit, die ich vorstellen möchte, ist das Proteinsprachenmodell ProTrek.
In der biologischen Forschung stehen viele Wissenschaftler vor diesem Problem: Sie verfügen über ein Genom mit vielen Proteinen, kennen jedoch deren spezifische Funktionen nicht.
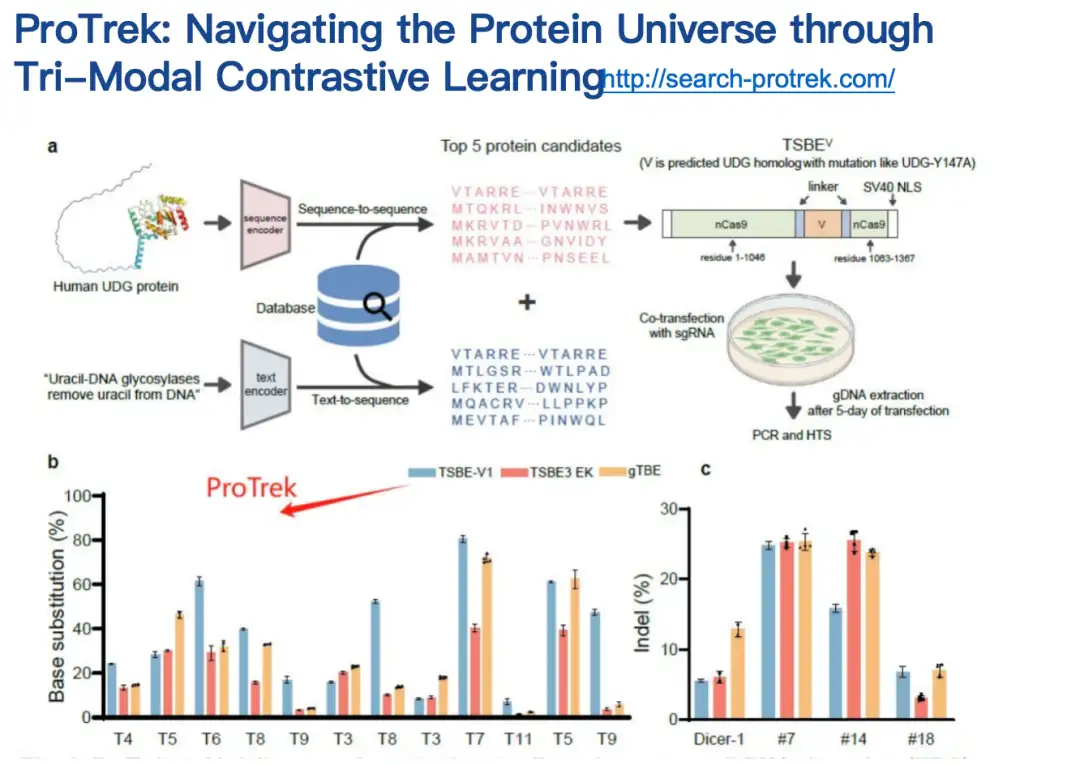
ProTrek ist ein trimodales Sprachmodell für kontrastives Lernen von Sequenz, Struktur und Funktion.Mit der Suchoberfläche in natürlicher Sprache können Benutzer den riesigen Proteinraum in Sekundenschnelle erkunden und nach Beziehungen zwischen allen paarweisen Kombinationen von Sequenz, Struktur und Funktion für neun verschiedene Aufgaben suchen. Mit anderen Worten: Mit ProTrek müssen Benutzer lediglich die Proteinsequenz eingeben und auf eine Schaltfläche klicken, um schnell Informationen zur Proteinfunktion und -struktur zu finden. Auf ähnliche Weise können Sie auch Sequenz- und Strukturinformationen basierend auf der Funktion und Sequenz- und Funktionsinformationen basierend auf der Struktur usw. finden. Darüber hinaus werden auch Sequenz-Sequenz- und Struktur-Struktur-Klassensuchen unterstützt.
ProTrek-Nutzungsadresse:
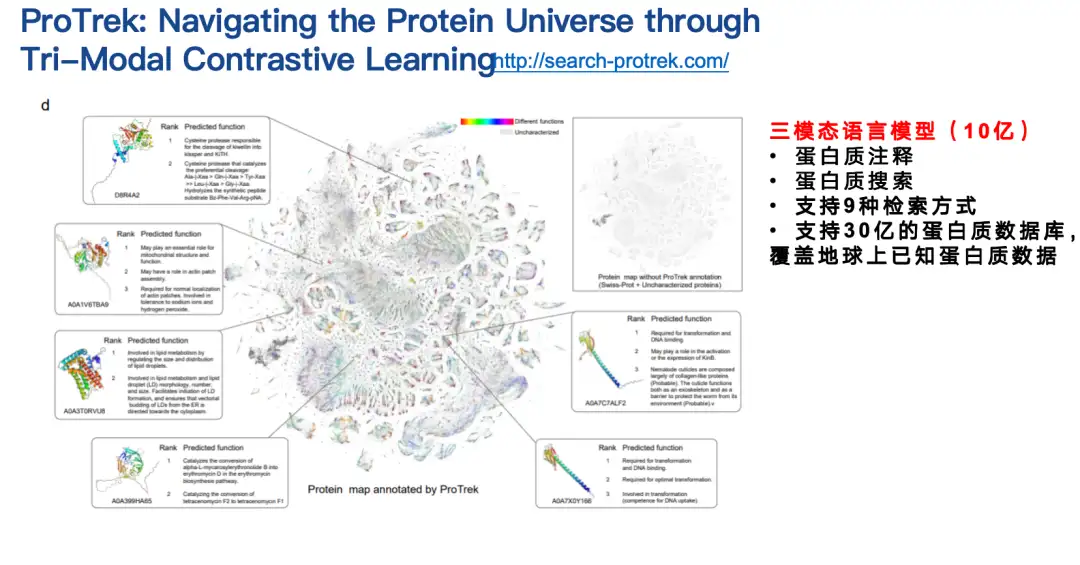
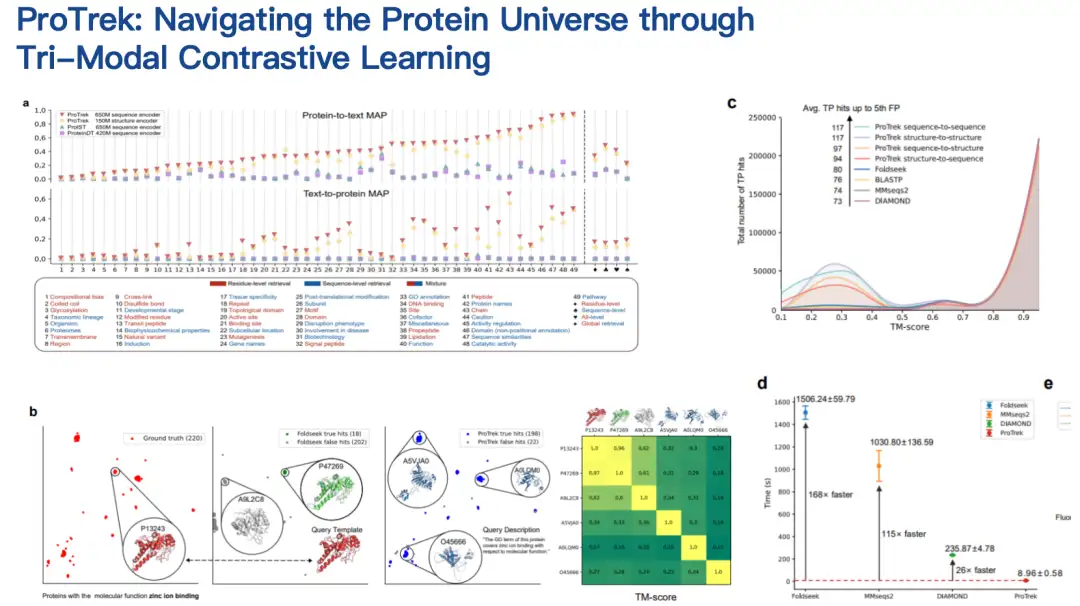
Unsere Mitarbeiter haben das ProTrek-Modell sowohl in Trocken- als auch in Nasstests bewertet.Im Vergleich zu bestehenden verwandten Methoden hat ProTrek erhebliche Leistungsverbesserungen erzielt. Darüber hinaus haben wir ProTrek verwendet, um eine große Menge an Daten für das Training unseres generativen Modells zu generieren, das ebenfalls gute Ergebnisse erzielte.
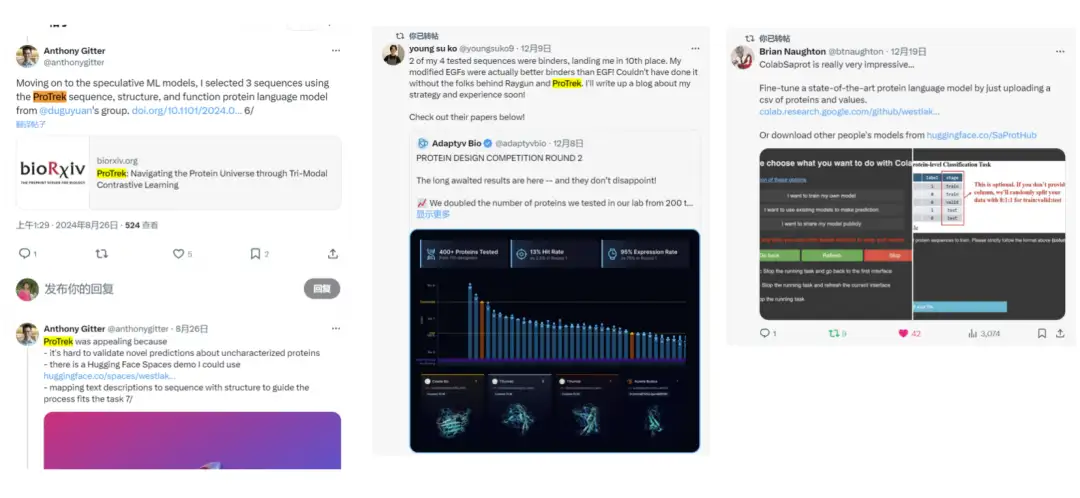
Wir haben auf Twitter bemerkt, dassViele Benutzer haben begonnen, ProTrek für Wettkämpfe zu verwenden.Darüber hinaus haben wir viel positives Feedback erhalten, was die Praxistauglichkeit des Modells zusätzlich unter Beweis stellt.
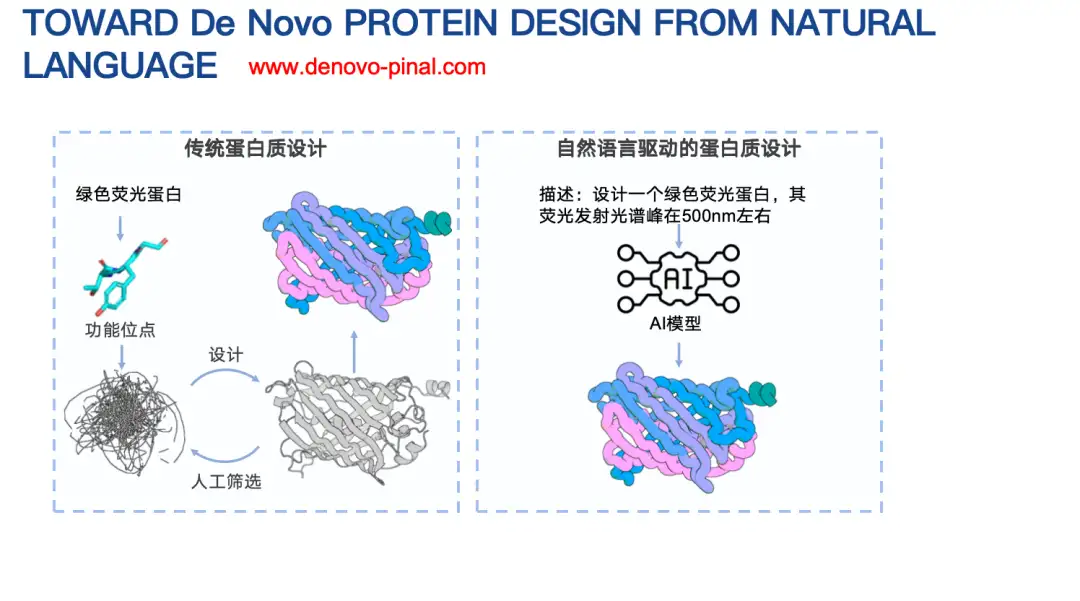
Pinal-Modell: Entwerfen Sie neue Proteinsequenzen durch einfache Texteingabe
Eine weitere unserer Arbeiten ist Pinal, ein Modell zum Design von Proteinen auf der Grundlage von Textbeschreibungen.
Beim traditionellen Proteindesign müssen normalerweise komplexe Faktoren berücksichtigt werden, beispielsweise Informationen zur Vorlage biophysikalischer Energiefunktionen. Da große Sprachmodelle bei vielen Aufgaben gute Ergebnisse erzielen, möchten wir untersuchen, ob es möglich ist, ein textbasiertes Proteinsprachenmodell zu entwerfen. In diesem Modell müssen wir lediglich die Informationen eines Proteins beschreiben, um seine Aminosäuresequenz zu entwerfen.
Pinal-Nutzungsadresse:
http://www.denovo-pinal.com/
Papieradresse:
https://www.biorxiv.org/content/10.1101/2024.08.01.606258v1
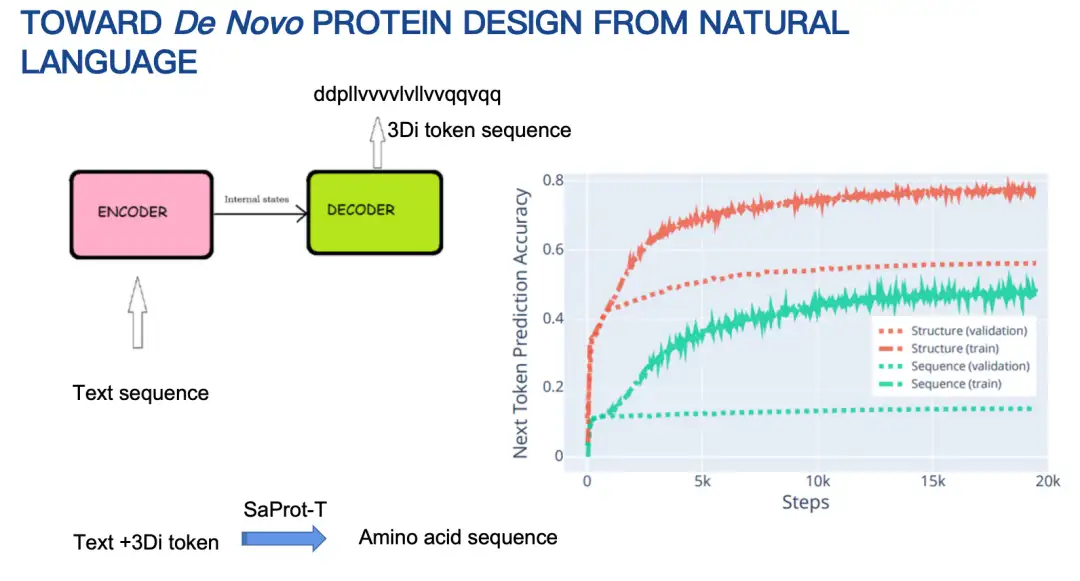
Lassen Sie mich kurz die Grundprinzipien von Pinal (16 Milliarden Parameter) vorstellen.Ursprünglich bestand unsere Idee darin, eine Encoder-Decoder-Architektur zu verwenden, die Text eingibt und eine Aminosäuresequenz ausgibt. Nach längerem Versuchen waren die Ergebnisse jedoch nicht optimal. Der Hauptgrund besteht darin, dass der Raum der Aminosäuresequenzen zu groß ist, was Vorhersagen erschwert.
Daher haben wir unsere Strategie angepasst, um zuerst die Proteinstruktur zu entwerfen und dann die Aminosäuresequenz basierend auf der Struktur und den Texthinweisen zu entwerfen. Auch hier wird die Proteinstruktur durch eine diskretisierte Kodierung dargestellt. Die Ergebnisse zeigen, dass die mit der Struktur kombinierte Entwurfsmethode hinsichtlich der Genauigkeit der nächsten Token-Vorhersage deutlich bessere Ergebnisse liefert als die Methode zur direkten Vorhersage der Aminosäuresequenz, wie in der folgenden Abbildung dargestellt.
Wir haben vor Kurzem von unseren Mitarbeitern eine Nasslaborbestätigung von Pinal erhalten.Pinal entwarf sechs Proteinsequenzen, von denen drei exprimiert wurden und bei zwei Sequenzen wurde eine entsprechende enzymkatalytische Aktivität nachgewiesen. Es ist erwähnenswert, dass wir in dieser Arbeit nicht Wert darauf gelegt haben, ein Protein zu entwickeln, das besser ist als der Wildtyp. Unser Hauptziel besteht darin, zu überprüfen, ob das auf Grundlage des Textes entworfene Protein die entsprechende Proteinfunktion besitzt.
Evolla-Modell: Die molekulare Sprache der Proteine entschlüsseln
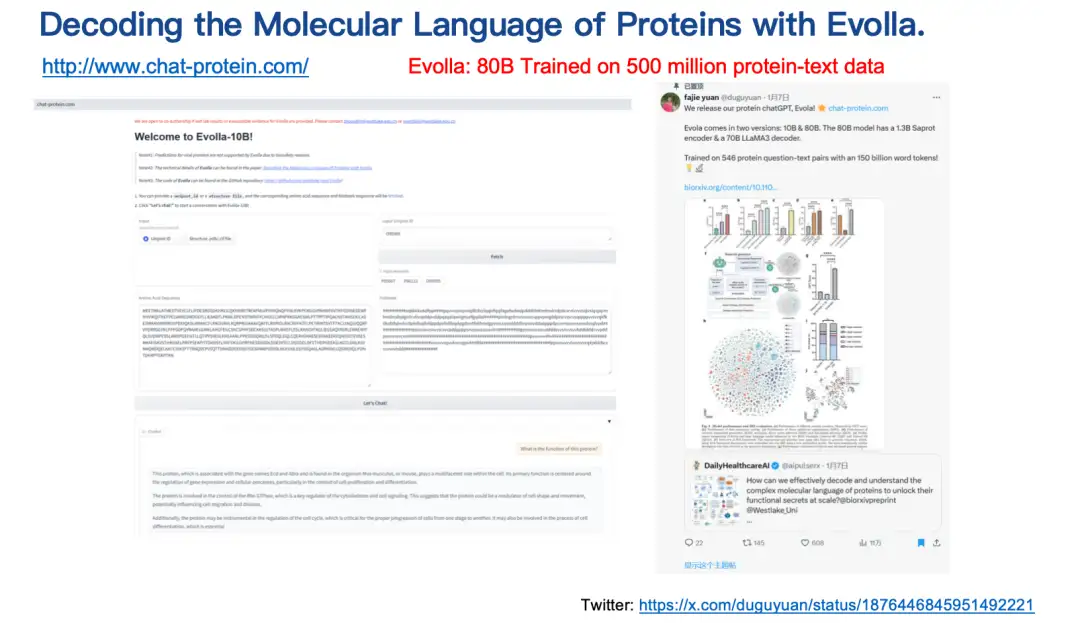
Das letzte vorgestellte Ergebnis ist das Evolla-Modell.Dabei handelt es sich um ein Modell zur Generierung einer Proteinsprache mit 80 Milliarden Parametern, eines der größten Open-Source-Modelle der Biologie, das zur Entschlüsselung der molekularen Sprache von Proteinen entwickelt wurde.
Durch die Integration von Proteinsequenz, Struktur und BenutzerabfrageinformationenEvolla liefert genaue Erkenntnisse zur Proteinfunktion.Benutzer müssen lediglich die Sequenz und Struktur des Proteins eingeben, dann Fragen stellen, beispielsweise zur Einführung in die Grundfunktion oder katalytische Aktivität des Proteins, und einfach auf eine Schaltfläche klicken. Evolla generiert dann eine detaillierte Beschreibung von etwa 200–500 Wörtern.
Evolla-Nutzungsadresse:
http://www.chat-protein.com/
Evolla-Papieradresse:
https://www.biorxiv.org/content/10.1101/2025.01.05.630192v2

Erwähnenswert ist, dass die für das Evolla-Projekt erforderlichen Trainingsdaten und die Rechenleistung enorm sind. Zwei unserer Doktoranden verbrachten fast ein Jahr allein mit der Erhebung und Verarbeitung der Trainingsdaten. Am Ende haben wir mithilfe synthetischer Daten mehr als 500 Millionen hochwertige Protein-Text-Paare generiert, die Hunderte Milliarden Wort-Token abdecken. Das Modell kann Enzymfunktionen recht genau vorhersagen.Es gibt jedoch unvermeidlich einige Illusionsprobleme.
Über das Team
Dr. Yuan Fajie von der Westlake University beschäftigt sich hauptsächlich mit angewandter wissenschaftlicher Forschung im Zusammenhang mit traditionellem maschinellem Lernen und interdisziplinären Themen und konzentriert sich auf die Erforschung großer KI-Modelle und der Computerbiologie. Er hat mehr als 40 wissenschaftliche Arbeiten in Top-Konferenzen und Fachzeitschriften im Bereich maschinelles Lernen und künstliche Intelligenz veröffentlicht (wie NeurIPS, ICLR, SIGIR, WWW, TPAMI, Molecular Cell usw.). Ausführliche Informationen zu den Teammitgliedern und Projektmitarbeitern finden Sie im Dokument.
Die Forschungsgruppe betreibt langjährige Forschung in den Bereichen maschinelles Lernen und KI + Bioinformatik. Sie können sich gerne für Doktoranden-, Wissenschaftliche Mitarbeiter-, Postdoktoranden- und Forscherstellen in der Forschungsgruppe bewerben. Studierende sind herzlich eingeladen, das Labor für Praktika zu besuchen. Interessenten können ihren Lebenslauf an [email protected] senden.








