Command Palette
Search for a command to run...
Das Team Der Universität Tokio Entwickelte Ein Deep-Learning-Framework Namens STAIG, Um Batch-Effekte Ohne Vorab-Ausrichtung Zu Eliminieren Und so Detaillierte Genetische Informationen Im Tumor-Mikroumfeld Zu enthüllen.

Biologische Gewebe sind komplexe Netzwerke aus mehreren Zelltypen, die durch spezifische räumliche Konfigurationen wichtige Funktionen erfüllen. In den letzten Jahren haben Fortschritte bei räumlichen Transkriptomik-Technologien (ST) wie 10x Visium, Slide-seq, Stereo-seq und STARmap es Biologen ermöglicht, genetische Daten innerhalb räumlicher Strukturen abzubilden und so tiefere Einblicke in verschiedene Krankheiten zu gewinnen.
Allerdings beruhen ST-Techniken in hohem Maße auf der Identifizierung räumlicher Regionen mit einheitlicher Genexpression und histologischen Merkmalen. derzeit,Es gibt zwei Hauptidentifizierungsmethoden: nicht-räumliches Clustering und räumliches ClusteringNicht-räumliche Clustering-Methoden führen das Clustering nur auf der Grundlage der Genexpression durch, was häufig zu inkohärenten Clustering-Ergebnissen führt. Methoden zur räumlichen Clusterbildung verwenden Graph-Convolution-Modelle, um Gen- und räumliche Informationen zu integrieren, verlassen sich jedoch bei der Konvertierung von ST-Daten in Graphstrukturen auf künstlich definierte Distanzstandards, was zu Verzerrungen führen kann. Gleichzeitig sind Methoden, die histologische Bilder verwenden, auch mit Herausforderungen verbunden, da sie anfällig für Veränderungen der Färbungsqualität sind. Darüber hinaus erfordert die Batch-Integration der meisten vorhandenen Methoden noch immer manuelle Eingriffe, wie etwa die manuelle Ausrichtung von Koordinaten oder die Nutzung zusätzlicher Tools.
Um diese Herausforderungen zu meistern,Ein Forschungsteam des Institute of Medical Science der Universität Tokio in Japan hat ein Deep-Learning-Framework namens STAIG (Spatial Transcriptomics Analysis with Image-Assisted Graph Comparative Learning) vorgeschlagen.Möglichkeit zur Integration von Genexpression, räumlichen Daten und histologischen Bildern ohne Ausrichtung.
STAIG extrahiert Merkmale aus mit Hämatoxylin und Eosin (H&E) gefärbten Bildern durch ein selbstüberwachtes Modell, ohne für das Vortraining auf umfangreiche histologische Datensätze angewiesen zu sein. Darüber hinaus passt STAIG die Graphstruktur während des Trainings dynamisch an und verwendet histologische Bildinformationen, um homologe negative Proben selektiv auszuschließen und so die durch die anfängliche Konstruktion verursachte Verzerrung zu reduzieren.
Schließlich identifiziert STAIG Gemeinsamkeiten in der Genexpression durch lokalen Vergleich, wodurch eine End-to-End-Batch-Integration ohne manuelle Koordinatenausrichtung ermöglicht und Batch-Effekte effektiv reduziert werden. Die Forscher bewerteten STAIG anhand mehrerer Datensätze.Die Ergebnisse zeigten, dass es eine gute Leistung bei der Identifizierung räumlicher Regionen bietet und detaillierte räumliche und genetische Informationen in der Tumormikroumgebung aufdecken kann, was das Verständnis komplexer biologischer Systeme fördert.
Die entsprechenden Ergebnisse wurden in Nature Communications unter dem Titel „STAIG: Spatial transcriptomics analysis via image-aided graph contrastive learning for domain exploration and alignment-free integration“ veröffentlicht.
Forschungshighlights:
* Das STAIG-Modell ermöglicht die Integration von Gewebeschnitten ohne Vorausrichtung und eliminiert Batch-Effekte
* Das STAIG-Modell ist auf Daten anwendbar, die von verschiedenen Plattformen erfasst wurden, unabhängig davon, ob histologische Bilder enthalten sind oder nicht.
* Forscher zeigen, dass STAIG räumliche Regionen mit hoher Genauigkeit identifizieren und neue Einblicke in die Tumormikroumgebung liefern kann, was sein breites Potenzial für die Analyse räumlicher biologischer Komplexität verdeutlicht.

Papieradresse:
https://www.nature.com/articles/s41467-025-56276-0
Downloadadresse des in dieser Studie verwendeten Datensatzes:
https://go.hyper.ai/m5YC4
Datensatz: Sammlung von ST-Datensätzen und Histologiebildern von verschiedenen Plattformen
Die Forscher luden öffentlich verfügbare ST-Datensätze und Histologiebilder von verschiedenen Plattformen herunter.Wie in der Abbildung unten gezeigt. Der ST-Datensatz umfasst den Datensatz zum dorsolateralen präfrontalen Kortex (DLPFC) des Menschen, den Datensatz zum Brustkrebs des Menschen, den Datensatz zum Maushirn, den Slide-seqV2-Datensatz, den STARmap-Datensatz usw.
Adresse zum Herunterladen des Datensatzes:
https://go.hyper.ai/m5YC4
* Der Datensatz des menschlichen dorsolateralen präfrontalen Kortex (DLPFC) von der 10x Visium-Plattform enthält 12 Scheiben von 3 Personen, von denen jede 4 Scheiben mit Intervallen von 10 μm und 300 μm bietet, und die Punktzahl jeder Scheibe reicht von 3.498 bis 4.789. Diese Scheiben wurden manuell als kortikale Schichten L1–L6 und weiße Substanz (WM) annotiert;
* Der Datensatz zum menschlichen Brustkrebs enthält 3.798 Punkte;
* Der Datensatz des Maushirns umfasst zwei Scheiben, eine vordere und eine hintere, mit 2.695 bzw. 3.355 Punkten;
* Für das Zebrafischmelanom analysierten die Forscher die Abschnitte A und B, die 2.179 bzw. 2.677 Flecken enthielten;
* Für das Ensemble-Experiment wurden DLPFC- und Mausgehirn-Datensätze verwendet. Der Stereo-seq-Datensatz des Riechkolbens der Maus enthält 19.109 Punkte mit einer Auflösung von 14 μm;
* Slide-seqV2-Datensatz mit einer Auflösung von 10 μm, einschließlich Hippocampus der Ratte (18.765 Punkte vom zentralen Viertelradius) und Bulbus olfactorius der Ratte (19.285 Punkte);
* Der STARmap-Datensatz enthält 1.207 Punkte;
* Für den MERFISH-Datensatz enthält das menschliche MTG 3.970 Punkte, während die VIS-Regionen von Maus 1 und Maus 2 5.995 bzw. 2.479 Punkte enthalten.
Modellarchitektur: Bildgestütztes Graphkontrastlernen für die räumliche Transkriptomikanalyse
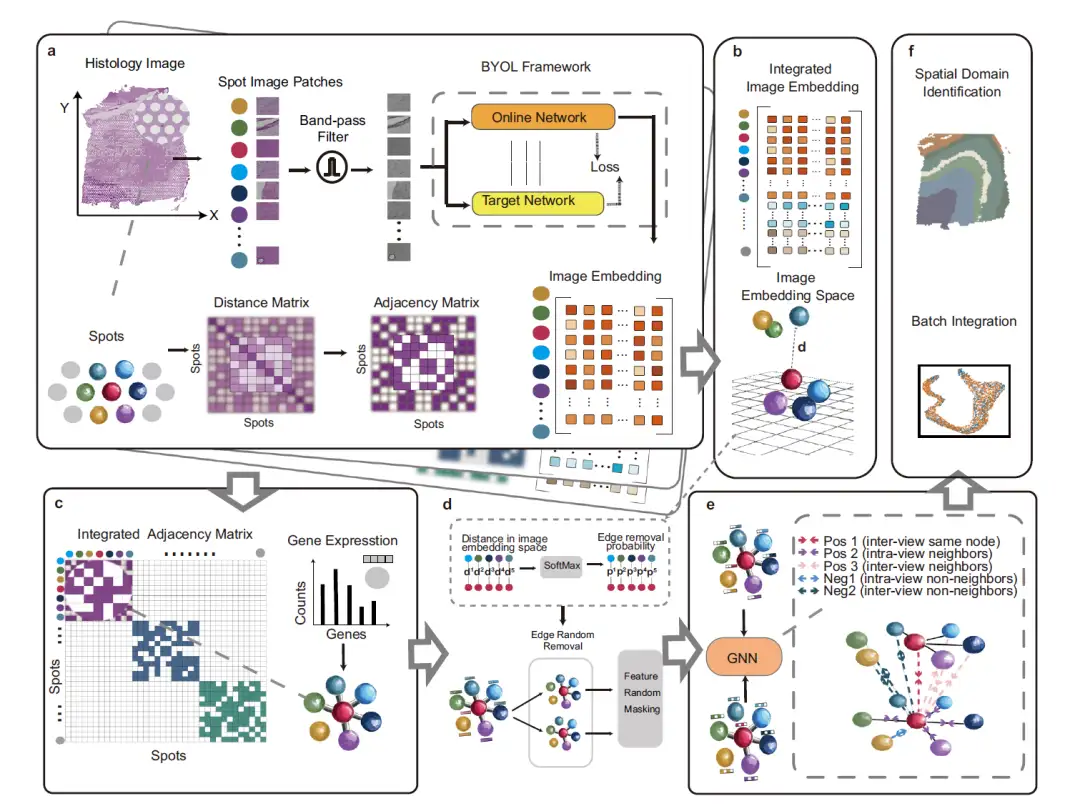
Die folgende Abbildung beschreibt das Gesamtframework von STAIG, einem Deep-Learning-Framework, das Graph-Kontrastlernen in Kombination mit leistungsstarker Merkmalsextraktion verwendet, um Genexpression, räumliche Koordinaten und histologische Bilder zu integrieren. Es besteht aus 6 Modulen:
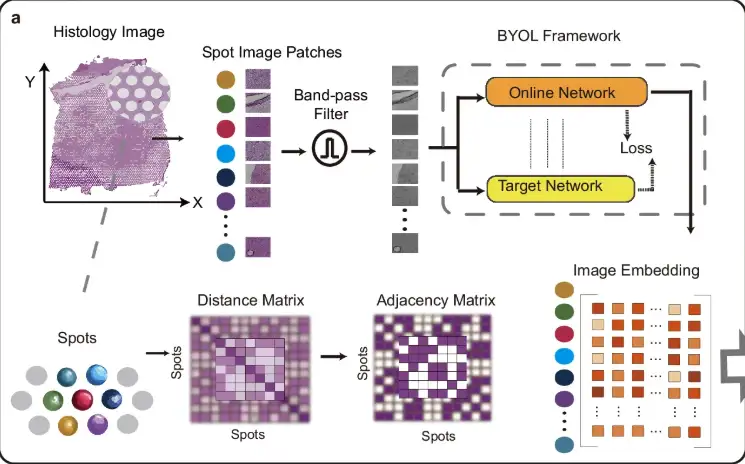
Um die Auswirkungen von Rauschen und ungleichmäßiger Gewebefärbung zu reduzieren, unterteilt STAIG, wie in Teil a der folgenden Abbildung gezeigt, zunächst das histologische Bild in kleine Patches (Spot Image Patches), die an den räumlichen Positionen der Datenpunkte ausgerichtet sind, und verwendet dann einen Bandpassfilter, um das Bild zu optimieren. Die Bildeinbettungsmerkmale werden durch das selbstüberwachte Modell Bootstrap Your Own Latent (BYOL) extrahiert und eine Adjazenzmatrix wird basierend auf der räumlichen Entfernung zwischen Datenpunkten erstellt.
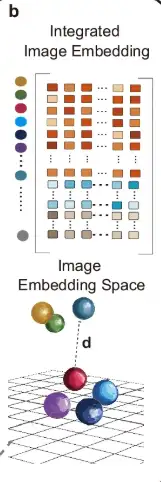
Wie in Teil b der folgenden Abbildung gezeigt, verwendet STAIG zur Integration der Daten verschiedener Gewebeschnitte eine vertikale Stapelmethode, um Merkmale mehrerer Gewebeschnitte einzubetten.
Wie in Teil c der folgenden Abbildung gezeigt, werden die Adjazenzmatrizen jedes Abschnitts mithilfe der diagonalen Platzierungsmethode zusammengeführt, um eine integrierte Adjazenzmatrix zu bilden, die dann zum Erstellen einer Graphstruktur mit Genexpressionsdaten als Knoteninformationen verwendet wird.
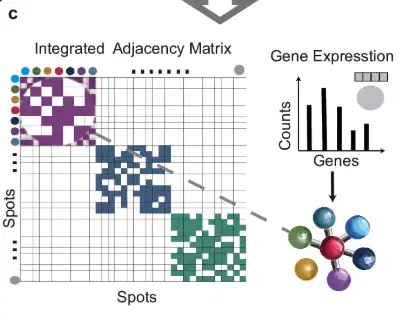
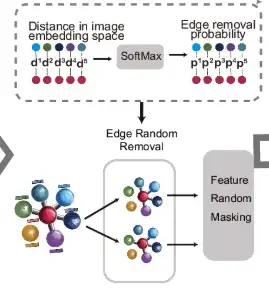
Wie in Teil d der folgenden Abbildung gezeigt, werden für die durch Kanten verbundenen Messpunkte ihre Abstände im Bildeinbettungsraum berechnet und die SoftMax-Funktion verwendet, um diese Abstände in die Wahrscheinlichkeit einer zufälligen Kantenentfernung umzuwandeln. Auf dieser Grundlage wird der ursprüngliche Graph zwei Runden zufälliger Kantenentfernung (Edge Random Removal) unterzogen, um zwei verbesserte Ansichten zu erzeugen. Anschließend werden die Knotenfunktionen in diesen Ansichten zufällig maskiert.
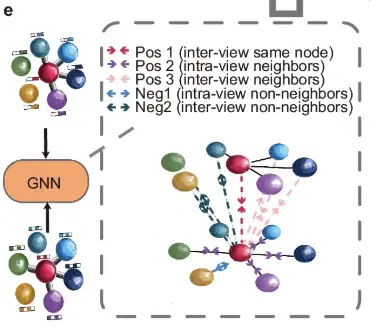
Anschließend wird die erweiterte Ansicht, wie in Teil e der Abbildung gezeigt, von einem gemeinsamen Graph-Neuralnetzwerk (GNN) verarbeitet und von einem benachbarten Kontrastziel geleitet, das darauf abzielt, benachbarte Knoten näher zusammenzubringen und nicht benachbarte Knoten in beiden Graphansichten weiter weg zu verschieben.
Schließlich generiert das trainierte GNN, wie in Abbildung f dargestellt, Einbettungen, um räumliche Regionen zu identifizieren und Batch-Effekte zwischen aufeinanderfolgenden Gewebeschnitten zu minimieren.

Forschungsergebnisse: STAIG zeigt überlegene Leistung unter verschiedenen Bedingungen
Das Forschungsteam führte eine umfassende Benchmark-Evaluierung durch, um STAIG mit anderen hochmodernen ST-Techniken zu vergleichen.Die Ergebnisse zeigen, dass STAIG unter verschiedenen Bedingungen eine überlegene Leistung zeigt.
Bewertung der Erkennungsleistung in Gehirnregionen
Um die Leistung von STAIG bei der Erkennung von Geweberegionen zu bewerten, verglichen die Forscher STAIG mit bestehenden Methoden, darunter Seurat, GraphST, DeepST, STAGATE, SpaGCN, SEDR, ConST, MuCoST und stLearn. Zu den Leistungsbewertungsindikatoren gehören:
* Adjusted Rand Index (ARI) und Normalized Mutual Information (NMI) (für manuell annotierte Datensätze).
* Silhouette-Koeffizient (SC) und Davis-Bolding-Index (DB) (für andere Datensätze).
① Leistung des menschlichen Gehirndatensatzes
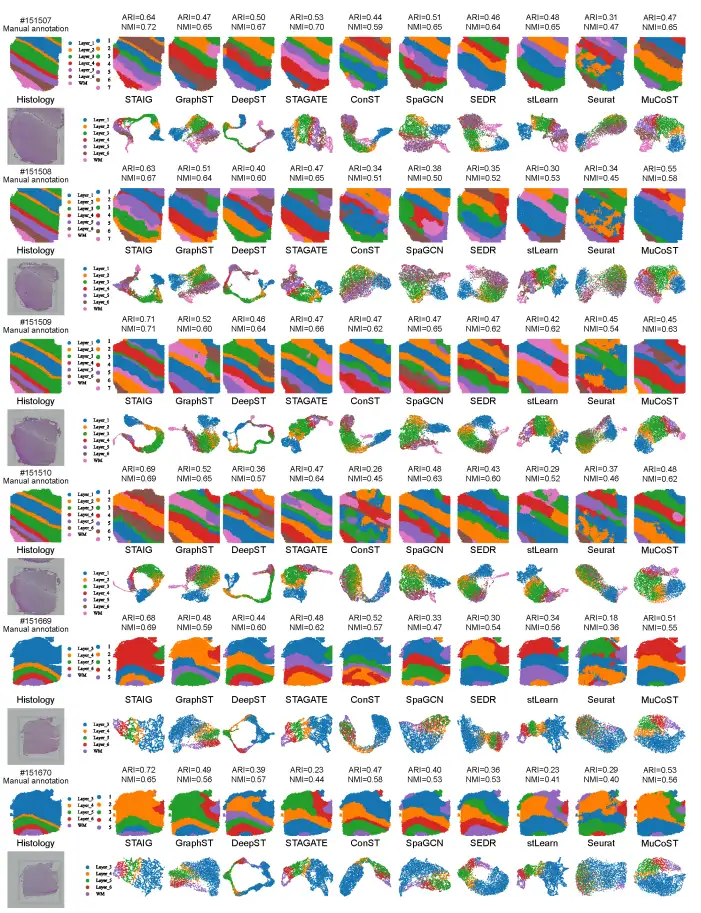
Gesamt,STAIG erzielt die beste Leistung bei Datensätzen des menschlichen Gehirns.Der höchste Medianwert von ARI (0,69) und NMI (0,71) wurde erreicht, wie in der folgenden Abbildung dargestellt:
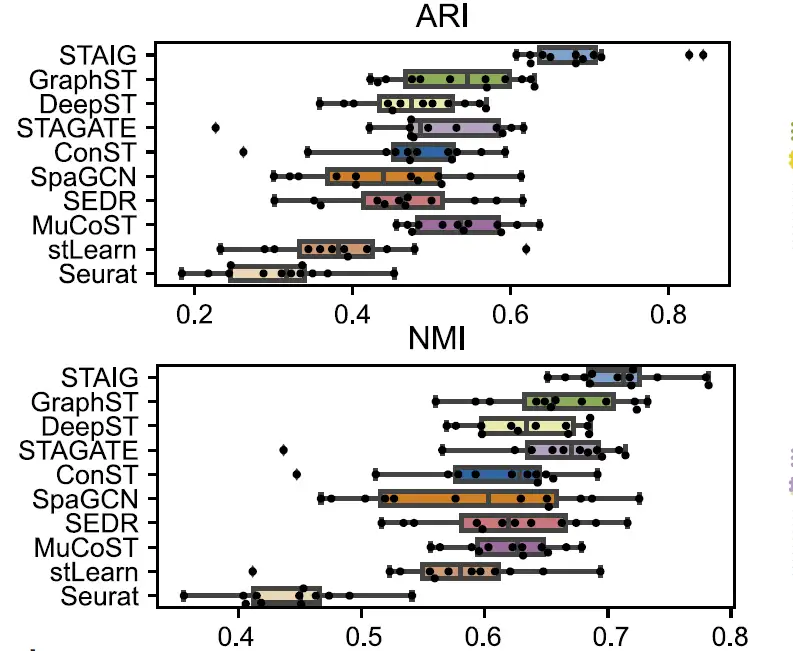
Im Vergleich dazu schneiden bestehende Methoden schlecht ab: stLearn schätzt einige Punkte falsch ein und übersieht einige Ebenen; GraphST hat einen ARI von 0,64 und einen NMI von 0,73, weist jedoch große Abweichungen bei den Positionen der Schichten L4 und L5 auf. Die ARIs anderer Methoden liegen zwischen 0,25 und 0,57 und die NMIs zwischen 0,42 und 0,69, was hauptsächlich auf eine ungenaue Identifizierung des Schichtverhältnisses zurückzuführen ist.
2 Leistung des Maushirn-Datensatzes
Wie in der Abbildung unten gezeigt, ist im Datensatz des MaushinterhirnsSTAIG konnte die Kleinhirnrinde und den Hippocampus erfolgreich identifizieren und darüber hinaus das Ammonshorn (CA) und den Gyrus dentatus unterscheiden.Sehr konsistent mit der Annotation des Allen-Maushirnatlas; trotz des Fehlens manueller Annotationen erreichte STAIG immer noch den höchsten SC (0,31) und den niedrigsten DB (1,11), was auf seine überlegene Clusterleistung hinweist.
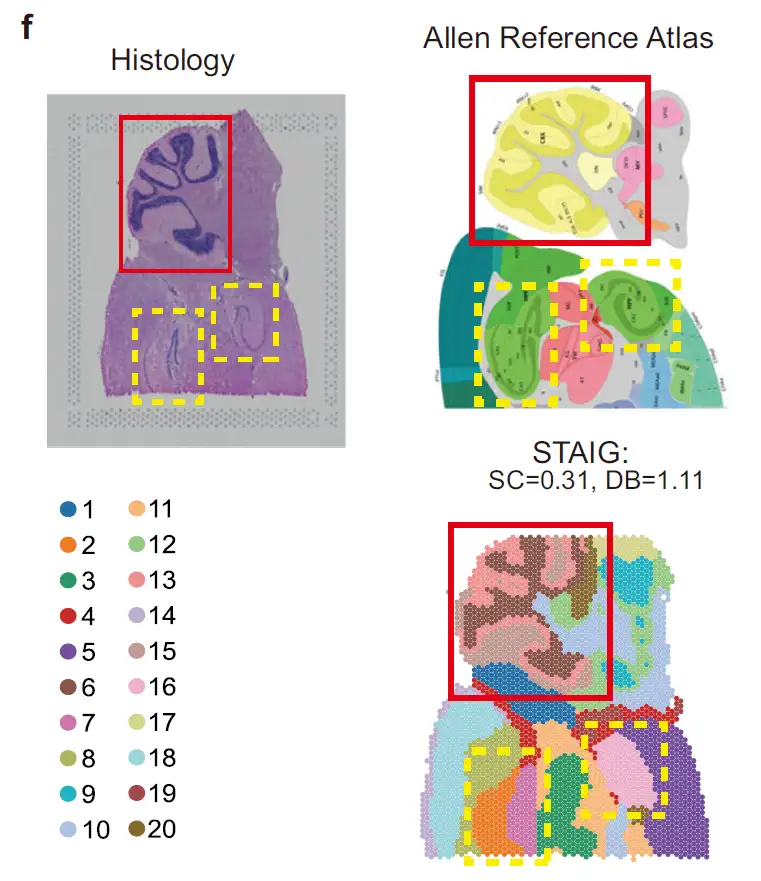
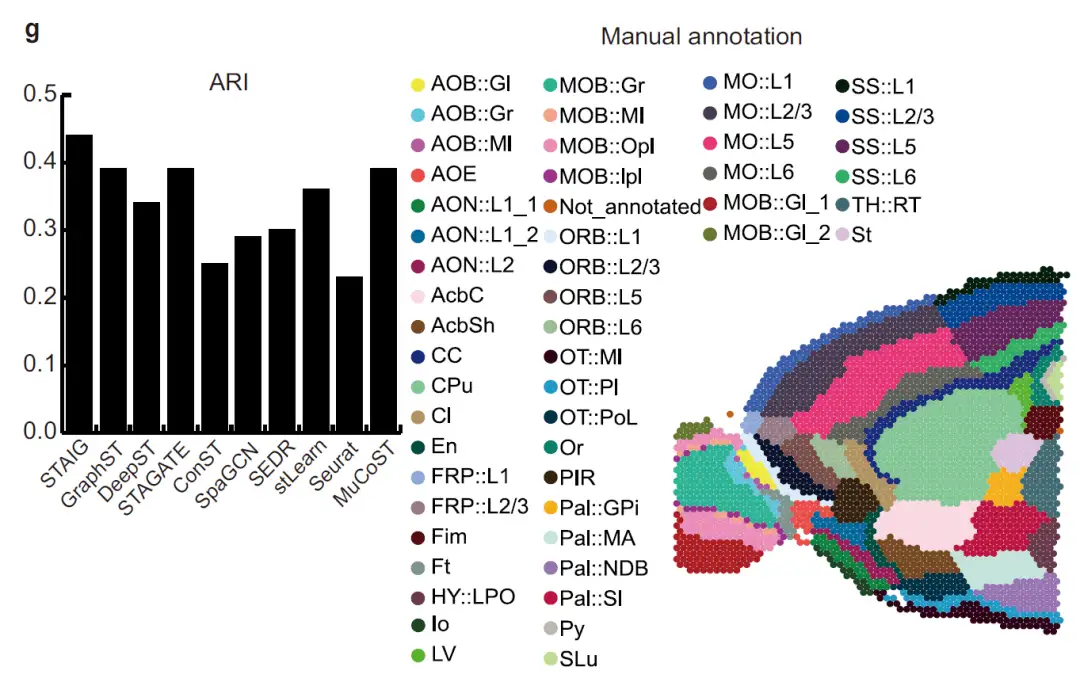
Wie in der Abbildung unten gezeigt, ist im Datensatz des Vorderhirns der MausSTAIG segmentierte den Bulbus olfactorius und das dorsale Pallium präzise.Nach Bezugnahme auf die manuelle Annotation von Long et al. erreichte sein ARI 0,44 und sein NMI 0,72, beides die höchsten Werte.
Effektivität der Bildmerkmalsextraktion
Um die Auswirkungen von Bildmerkmalen zu untersuchen, verwendeten die Forscher den KNN-Algorithmus, um die von STAIG extrahierten Bildmerkmale mit denen zu vergleichen, die mit anderen Methoden (stLearn, DeepST und ConST) extrahiert wurden.
① Analyse von Hirngewebeschnitten
Am Beispiel von Abschnitt #151507, wie in der folgenden Abbildung gezeigt, werden die Bildmerkmale von stLearn stark durch die Färbungsintensität beeinträchtigt, was zu einer Nichtübereinstimmung mit der tatsächlichen hierarchischen Annotation führt. Obwohl DeepST und ConST Deep Learning verwenden, gelingt es ihnen nicht, die komplexen Texturmerkmale des Hirngewebes genau zu erfassen. Die Ergebnisse der Merkmalsextraktion von STAIG stimmen weitgehend mit den manuell annotierten Ebenen überein. Obwohl einige Grenzen noch leicht verschwommen sind, werden sie durch Färbungsunterschiede kaum beeinflusst.

2 Bildanalyse von Brustkrebsgewebe
Die Forscher testeten die Fähigkeit zur Bildmerkmalsextraktion außerdem mithilfe von H&E-gefärbten Bildern von menschlichem Brustkrebs, wie in der folgenden Abbildung dargestellt.
Die Ergebnisse zeigten, dass das Bild von stLearn gemischte Tumore und normale Bereiche mit schlechter Unterscheidung aufweist. ConST schien das Bild in verschiedene Regionen zu unterteilen, aber nach dem Vergrößern wichen die Regionsgrenzen stark von den manuellen Anmerkungen ab; DeepST konnte keine effektiven Bildmerkmale extrahieren;STAIG identifiziert Tumorregionen präzise. Die Ergebnisse der räumlichen Clusterung weisen ein hohes Maß an regionaler Kohärenz auf und die segmentierten Regionen stimmen nahezu perfekt mit den manuell annotierten Konturen überein.Die hervorragende Fähigkeit zur Bildmerkmalsextraktion wurde bestätigt.

Definition der Tumormikroumgebung bei menschlichem Brustkrebs ST
Bei der Analyse eines Datensatzes zum Thema Brustkrebs beim MenschenDie Forscher stellten fest, dass die Ergebnisse von STAIG in hohem Maße mit manuellen Anmerkungen übereinstimmten und den höchsten ARI (0,64) und NMI (0,70) erreichten.Es ist bemerkenswert, dass STAIG eine etwas andere, aber verfeinerte räumliche Schichtung vorschlägt, insbesondere für die manuell annotierte Region Healthy_1 (Abbildung 2a), die STAIG in die Untercluster 3 und 4 unterteilt (Abbildung 2b).
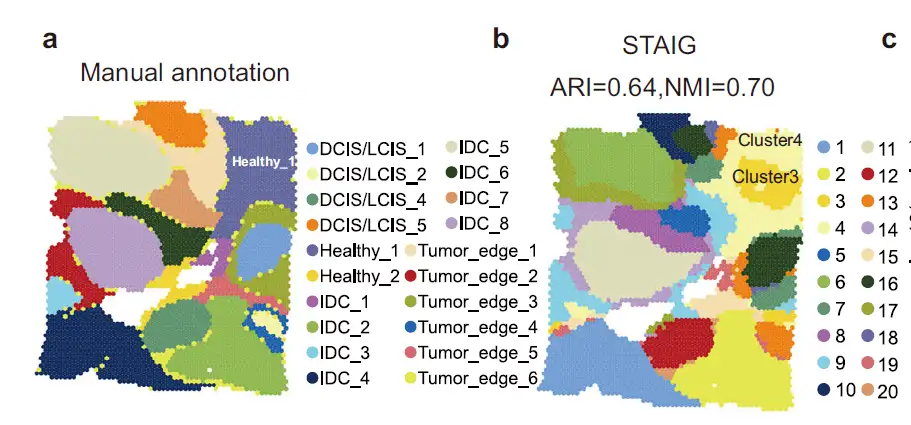
Zusammenfassend stellten wir durch die multimodale Integration von STAIG fest, dass Subcluster 3 eine CAF-dichte Tumormikroumgebung bildete und die molekularen Eigenschaften CAF-reicher Bereiche offenbarte.
Deep Learning bietet leistungsstarke Tools für die ST-Technologieentwicklung
Durch die rasante Entwicklung der Genomik und der ST-Technologie ist die biomedizinische Gemeinschaft in der Lage, die räumliche Verteilung der Genexpression innerhalb von Geweben zu erforschen und so die komplexen Funktionen und Strukturen von Organismen aufzudecken. Die ST-Technologie liefert nicht nur quantitative Informationen zur Genexpression, sondern bewahrt auch die räumliche Beziehung der Zellen in Geweben, sodass Forscher das Gewebemikroumfeld, Zellinteraktionen und die räumlichen Merkmale der Krankheitsentwicklung untersuchen können. Jedoch,Da ST-Daten normalerweise Probleme wie hohe Dimensionalität, starkes Rauschen und Batch-Effekte aufweisen, ist die effektive Integration und Analyse dieser Daten zu einer zentralen Herausforderung in der aktuellen Forschung geworden.
Die Einführung von Deep-Learning-Techniken, insbesondere Graph Neural Networks (GNNs) und kontrastiven Lernmethoden, bietet leistungsstarke Tools für die Analyse von ST-Daten. Herkömmliche Analysemethoden basieren häufig auf Dimensionsreduzierung und Clustering, während Deep-Learning-Methoden mehrstufige Merkmale automatisch extrahieren und die Datendarstellung durch End-to-End-Training optimieren können. Wie oben erwähnt, kann die GNN-basierte Methode räumliche Nachbarschaftsinformationen verwenden, um eine Graphstruktur zu erstellen, sodass das Modell nicht nur die Genexpression erfassen, sondern auch die räumlichen Abhängigkeiten zwischen Zellen erlernen kann. Durch die Einführung des kontrastiven Lernens wird die Generalisierungsfähigkeit des Modells weiter verbessert, sodass es wichtige räumliche Merkmale ohne Annotation erlernen kann.
Darüber hinaus hat die Branche auch bei der Kombination von Deep Learning und ST-Technologie große Fortschritte gemacht:
November 2024Das von Yang Yungui vom National Center for Bioinformatics of China geleitete Team und das von Zhang Shihua vom Institute of Mathematics and Systems Science der Chinesischen Akademie der Wissenschaften geleitete Team haben ein auf Deep Learning basierendes räumliches Transkriptom-Zellannotation-Tool namens STASCAN entwickelt.Durch die Integration von Genexpressionsprofilen und dem Lernen von Zellmerkmalen aus histologischen Bildern sagen wir Zelltypen in unbekannten Bereichen von Gewebeschnitten voraus und kommentieren Zellen innerhalb des erfassten Bereichs, wodurch wir die räumliche Zellauflösung erheblich verbessern. Darüber hinaus ist STASCAN auf verschiedene Datensätze aus unterschiedlichen ST-Techniken anwendbar und bietet erhebliche Vorteile bei der Entschlüsselung der hochauflösenden Zellverteilung und der Auflösung einer verbesserten Gewebearchitektur.
Dieses Ergebnis wurde in Genome Biology unter dem Titel „STASCAN entschlüsselt hochaufgelöste Zellverteilungskarten in der räumlichen Transkriptomik durch Deep Learning“ veröffentlicht.
* Papieradresse:
https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03421-5
23. Januar 2025Ein Forschungsteam der Princeton University in den USA hat einen neuen Deep-Learning-Algorithmus namens GASTON (Gradient Analysis of Spatial Transcriptomics Organization with Neural Networks) entwickelt. Durch die Kombination unbeaufsichtigter tiefer neuronaler Netzwerke mit interpretierbaren Algorithmen schlug GASTON auf innovative Weise das Konzept der „Isotiefe“ vor, das der Höhe auf einer topografischen Karte ähnelt und zur Quantifizierung der räumlichen topologischen Struktur der Genexpression in Gewebeschnitten verwendet wird. ,
Durch Isotiefe und ihren Gradienten können Forscher nicht nur verschiedene räumliche Bereiche des Gewebes segmentieren, sondern auch die kontinuierlichen Veränderungstrends und Schlüsselmarkergene der Genexpression innerhalb des Gewebes identifizieren. Die Studie demonstrierte die erfolgreiche Anwendung von GASTON in einer Vielzahl biologischer Proben, darunter Mäusehirn, Riechkolben von Mäusen, Mikroumgebung von Dickdarmkrebstumoren usw. Die Ergebnisse zeigen, dass GASTON die Gewebestruktur genau analysieren, die räumliche Verteilung und Änderungsmuster von Zelltypen aufdecken und viele räumliche Genexpressionsmuster entdecken kann, die von anderen Methoden übersehen werden.
Die entsprechenden Ergebnisse wurden in Nature Methods unter dem Titel „Mapping the topography of spatial gene expression with interpretable deep learning“ veröffentlicht.
* Papieradresse:
https://www.nature.com/articles/s41592-024-02503-3
Offensichtlich verbessert die Kombination aus Deep Learning und ST-Technologie nicht nur die Fähigkeit zur Datenintegration und Rauschunterdrückung, sondern fördert auch die eingehende Auswertung räumlicher biologischer Informationen. Mit der Zunahme der Rechenressourcen und der Optimierung der Algorithmen wird Deep Learning in Zukunft eine wichtigere Rolle bei der ST-Datenanalyse spielen und die Präzisionsmedizin und personalisierte Behandlung stärker unterstützen.
Quellen:
1.https://static-content.springer.com/esm/art%3A10.1038%2Fs41467-025-56276-0/MediaObjects/41467_2025_56276_MOESM1_ESM.pdf
2.https://www.bjqykxy.com/kexueyanjiu/dongwuzhiwu/7361.html
3.https://news.qq.com/rain/a/20250128A057OQ00?suid=&media_id=
4.https://www.medsci.cn/article/show_








