Command Palette
Search for a command to run...
Das Team Von Zhang Yang an Der National University of Singapore Entwickelte Einen Algorithmus Zur Vorhersage Der RNA-Struktur Der Zweiten Generation, Der SOTA in Mehreren Benchmarktests Übertraf

Das Verständnis der Struktur und Funktion von RNA-Molekülen war schon immer eine zentrale Forschungsrichtung in der Molekularbiologie und der Pharmaindustrie. RNA, insbesondere nichtkodierende RNA (ncRNA), kann sich in spezifische Strukturen falten und spielt eine wichtige Rolle in verschiedenen zellulären Prozessen wie der Genregulation (wie Transkription und Translation), Katalyse, biologischen Signaltransduktion und Stressreaktion.
Mit der rasanten Entwicklung der Hochdurchsatz-Sequenzierungstechnologie sind die RNA-Sequenzdaten exponentiell gewachsen, doch die Lücke zwischen bekannten Sequenzen und experimentell aufgelösten RNA-Strukturen wird immer größer. Daher wird es immer dringlicher, die atomare Struktur der RNA ausschließlich auf Grundlage ihrer Rohsequenz aufzuklären. Forscher haben eine Vielzahl von Methoden zur Untersuchung der RNA-Struktur entwickelt, darunter strukturbiologische Techniken wie Röntgenkristallographie, Kernspinresonanzspektroskopie und Kryoelektronenmikroskopie (Kryo-EM). Obwohl diese experimentellen Techniken eine höhere Auflösung bieten können, ist die experimentelle Aufklärung der dreidimensionalen Struktur von RNA oft kostspielig und in manchen Fällen schwierig zu erreichen. daher,Es besteht eine wachsende Nachfrage nach rechnergestützten Methoden zur Vorhersage der dreidimensionalen Struktur hochwertiger RNA direkt aus der Sequenz.
Unter „Ab initio RNA-Strukturvorhersage“ versteht man eine Methode, die die dreidimensionale Struktur von RNA direkt aus ihrer Sequenz vorhersagt, ohne auf experimentelle Daten oder Vorkenntnisse zurückzugreifen. Der Kern dieser Methode besteht darin, Computersimulationen und Techniken der Computerchemie zu verwenden, um die dreidimensionale Konformation von RNA-Molekülen durch mathematische Modelle und Algorithmen vorherzusagen.
Die neuesten Forschungsergebnisse des Teams von Professor Zhang Yang an der National University of Singapore haben die „Ab-initio-RNA-Strukturvorhersage“ auf ein höheres Niveau gehoben.Forscher schlugen ein hochpräzises Framework zur Vorhersage der RNA-Struktur auf Basis von Deep Learning vor: DRfold2.Es integriert ein vortrainiertes RNA-Composite-Language-Modell (RCLM) und ein Denoising-Strukturmodul für die End-to-End-RNA-Strukturvorhersage. DRfold2 schneidet sowohl bei der Vorhersage der globalen Topologie als auch der Sekundärstruktur im Vergleich zu anderen hochmodernen Methoden bei mehreren Benchmarks gut ab.
Eine detaillierte Analyse zeigt, dass diese Verbesserung hauptsächlich auf die Fähigkeit von RCLM zurückzuführen ist, koevolutionäre Muster zu erfassen und auf seinen effizienten Rauschunterdrückungsprozess.Dies verbessert die Genauigkeit der unbeaufsichtigten Kontaktvorhersage von DRfold2 im Vergleich zu bestehenden Methoden um mehr als 100%.
Die entsprechenden Ergebnisse wurden auf der Preprint-Plattform bioRxiv unter dem Titel „Ab initio RNA structure prediction with composite language model and denoised end-to-end learning“ veröffentlicht.
Forschungshighlights:
* DRfold2 integriert ein vortrainiertes RNA Composite Language Model (RCLM) und ein Denoising-Strukturmodul für die End-to-End-RNA-Strukturvorhersage
* Durch eine einzigartige Kombination aus zusammengesetzter Sprachmodellierung, auf Rauschunterdrückung basierendem End-to-End-Lernen und Deep-Learning-gesteuerter Nachoptimierung eröffnet DRfold2 eine neue Richtung für die „Ab-initio-RNA-Strukturvorhersage“.
* DRfold2 ergänzt AlphaFold3 optimal und erzielt nach der Integration in das Optimierungsframework statistisch signifikante Genauigkeitsverbesserungen.

Papieradresse:
https://www.biorxiv.org/content/10.1101/2025.03.05.641632v1
Laden Sie den DRfold2-RNA-Strukturtestdatensatz herunter:
Datensatz: Erstellen Sie einen unabhängigen Testdatensatz
Um die Leistung von DRfold2 objektiv zu bewerten,Die Forscher erstellten einen unabhängigen Testdatensatz mit 28 RNA-Strukturen.Ihre Sequenzlängen betragen alle weniger als 400 nts und stammen aus den folgenden drei Kategorien:
* Neueste RNA-Puzzles Zielsequenzen
* RNA-Zielsequenzen im CASP15-Wettbewerb
* Die zuletzt veröffentlichten RNA-Strukturen in der Protein Data Bank (PDB)-Datenbank mit Stand vom 1. August 2024
Bemerkenswerterweise schlossen die Forscher große synthetische RNA-Strukturen aus dem CASP15-Datensatz aus, da diese von in der Natur vorkommenden RNA-Strukturen abweichen, die im Mittelpunkt der Funktionsanalyse und des Arzneimitteldesigns stehen.
Um eine strenge Modellbewertung zu gewährleisten, enthält der Trainingssatz nur RNA-Strukturen, die vor 2024 veröffentlicht wurden, und schließt RNAs mit einer Sequenzähnlichkeit von mehr als 80% zum Testdatensatz aus.
Laden Sie den DRfold2-RNA-Strukturtestdatensatz herunter:
Modellarchitektur: eine neue Pipeline zur Vorhersage von RNA-3D-Strukturen DRfold2
DRfold2 ist eine neue Pipeline zur Vorhersage von RNA-3D-Strukturen, die aus vier Kernmodulen besteht: (1) RNA Composite Language Model (RCLM), (2) RNA Transformer Block, (3) Denoising Structure Module und (4) endgültige Modellauswahl und -optimierung durch das CSOR-Protokoll, wie in Abbildung A unten dargestellt:
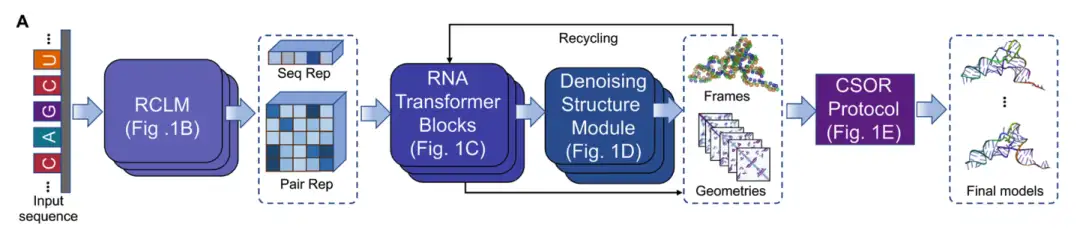
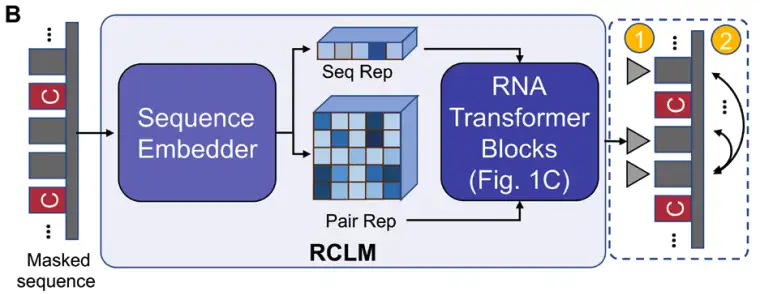
Ausgehend von einer RNA-Sequenz,DRfold2 codiert zunächst die Abfragesequenz mithilfe eines vortrainierten RNA-Composite-Language-Modells (RCLM).Generieren Sie eine Sequenzdarstellung (Seq Rep) und eine Paardarstellung (Pair Rep). RCLM wird mithilfe der Methode der zusammengesetzten Wahrscheinlichkeitsmaximierung anhand großer, unbeaufsichtigter Sequenzdaten trainiert, um eine effizientere Sequenzmustererkennung zu erreichen, wie in Abbildung B unten dargestellt:
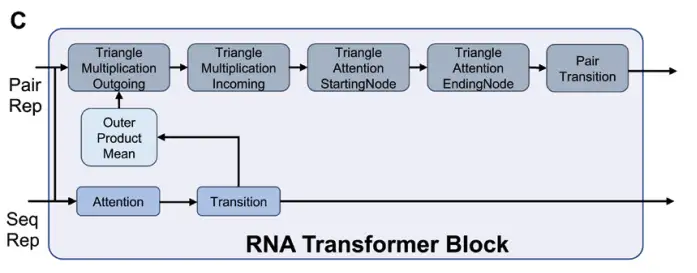
Diese Sequenzen und gepaarten Darstellungen werden dann zur Verarbeitung in das RNA-Transformer-Modul eingegeben, um die für die Faltung der RNA-Struktur erforderlichen Schlüsselmerkmalsdarstellungen zu generieren, wie in Abbildung C unten dargestellt:
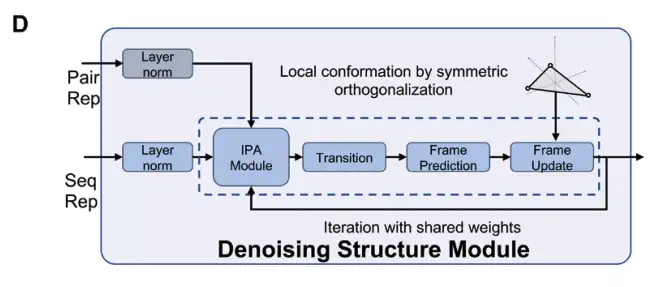
Als nächstes verwendet DRfold2 das Denoised RNA Structure Module (DRSM), um RNA-Konformationen in einer End-to-End-Weise zu generieren, wie in Abbildung D unten dargestellt:
Das endgültige RNA-Strukturmodell wird durch das CSOR-Nachbearbeitungsprotokoll geprüft und optimiert, um aus den an mehreren Kontrollpunkten generierten Konformationen das beste Modell auszuwählen und zu verfeinern, wie in Abbildung E unten dargestellt:
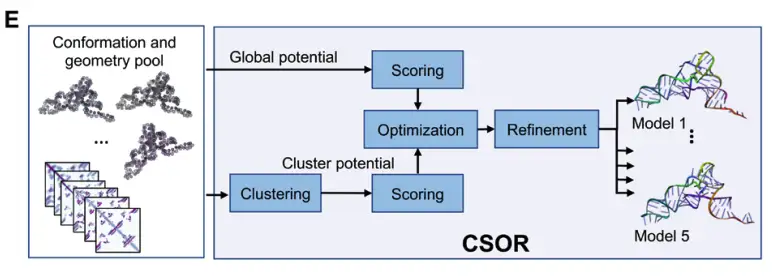
Obwohl DRfold2 einen ähnlichen Namen wie die frühere DRfold-Methode des Teams trägt, führt es bedeutende Fortschritte ein, die auf einem völlig anderen Framework basieren.Das Wichtigste ist die Integration eines zusammengesetzten Sprachmodells, das die Fähigkeit zur Darstellung von RNA-Sequenzen und -Paaren erheblich verbessert.Darüber hinaus integriert die Vorhersage-Pipeline ein Denoising-RNA-Strukturmodul (DRSM), das eine kontrollierte Störungsstrategie verwendet, um Strukturtransformationen durch effiziente Korrektur verrauschter RNA-Konformationen robust zu erlernen.
Die Forscher haben den DRfold2-Onlineserver und den lokalen Code öffentlich zugänglich gemacht unter:
https://zhanglab.comp.nus.edu.sg/DRfold2
Forschungsergebnisse: DRfold2 übertrifft andere hochmoderne Methoden bei mehreren Benchmarks
Die Forscher verglichen DRfold2 zunächst mit fünf hochmodernen Methoden zur Vorhersage der RNA-Struktur, darunter RNAComposer (basierend auf Fragmentassemblierung und -optimierung), trRosettaRNA (Deep-Learning-Methode), RhoFold (End-to-End-Deep-Learning-Methode), RoseTTAFoldNA (End-to-End-Deep-Learning-Methode) und DeepFoldRNA (Deep-Learning-Methode).
Wie in der folgenden Abbildung dargestellt, verglichen die Forscher die TM-Score- und RMSD-Auswertungsergebnisse von DRfold2 und der Benchmark-Methode bei verschiedenen Schwellenwerten für die Sequenzähnlichkeit (50%-80%). Unter diesen ist der TM-Score eine längenunabhängige Bewertungsfunktion, die zur Beurteilung der Gesamtqualität der vorhergesagten RNA-Struktur verwendet wird. Der Wertebereich ist 0-1. Je höher der Wert, desto größer ist die Ähnlichkeit zwischen der vorhergesagten Struktur und der tatsächlichen Struktur.
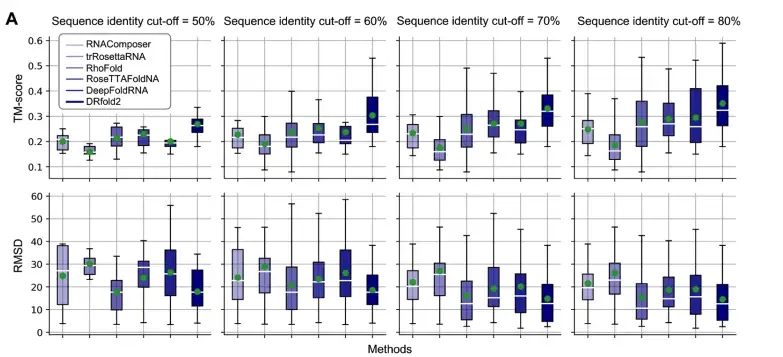
Die Ergebnisse zeigen, dass DRfold2 unter allen Schwellenwerten für die Sequenzähnlichkeit immer den höchsten durchschnittlichen TM-Score erzielt.Zum Beispiel:
* Unter dem Ähnlichkeitsschwellenwert von 80% beträgt der durchschnittliche TM-Score von DRfold2 0,351 und ist damit 18,6% höher als beim zweitplatzierten DeepFoldRNA (TM-Score = 0,296).
* Unter dem Ähnlichkeitsschwellenwert von 50% (dem strengsten Testsatz) kann DRfold2 immer noch einen durchschnittlichen TM-Score von 0,269 erreichen, der 17,5% höher ist als beim zweitplatzierten RoseTTAFoldNA (TM-Score = 0,229).
* Darüber hinaus ist die RMSD (Root Mean Square Deviation) von DRfold2 bei allen Schwellenwerten der Sequenzähnlichkeit immer niedriger als bei allen Kontrollmethoden, was darauf hindeutet, dass die vorhergesagte Struktur näher an der tatsächlichen RNA-Struktur liegt.
Die Forscher verwendeten außerdem das HDV-ähnliche Ribozym CPEB3 des Schimpansen (PDB-ID: 7QR3) als Beispiel. Die RNA ist 69 Nukleotide lang und die Vorhersageeffekte verschiedener Methoden auf ihre RNA-Tertiärstruktur wurden analysiert. Die Ergebnisse sind wie folgt:
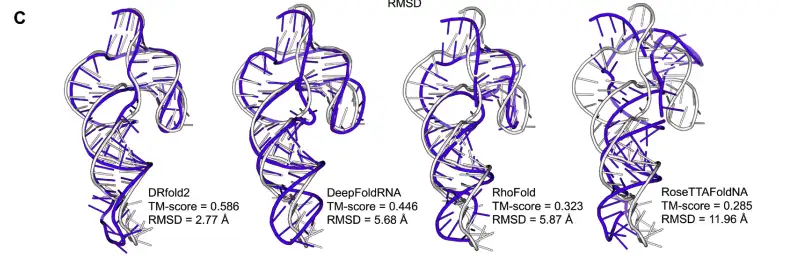
* DRfold2 hat die gesamte topologische Struktur des Ribozyms mit einem TM-Score von 0,586 und einem RMSD von nur 2,77 Å genau erfasst.
* DeepFoldRNA zeigt hinsichtlich der gesamten helikalen Anordnung eine gute Leistung, die Richtung der Haarnadelschleife weicht jedoch erheblich ab, was zu einem RMSD von bis zu 5,68 Å führt, was der doppelten Abweichung von DRfold2 entspricht.
* RhoFold und RoseTTAFoldNA weisen größere Fehler bei der räumlichen Vorhersage in Verbindungsbereichen auf, wodurch der TM-Score auf 0,323 bzw. 0,285 sinkt.
* Die höchste Sequenzähnlichkeit zwischen der Ziel-RNA und dem Trainingsdatensatz beträgt nur 60,9%, was darauf hindeutet, dass DRfold2 auch ohne homologe Vorlagen zuverlässige Strukturvorhersagen für neue RNA-Sequenzen liefern kann.
Diese Ergebnisse zeigen, dass:Die umfassende probabilistische Darstellung, die durch Sprachmodelle höherer Ordnung wie RCLM bereitgestellt wird, verbessert die Fähigkeit, gemeinsam entstehende Muster und räumliche Einschränkungen zu erlernen, erheblich.Somit wurde durch das End-to-End-Netzwerk von DRfold2 eine genauere 3D-RNA-Strukturmodellierung erreicht.
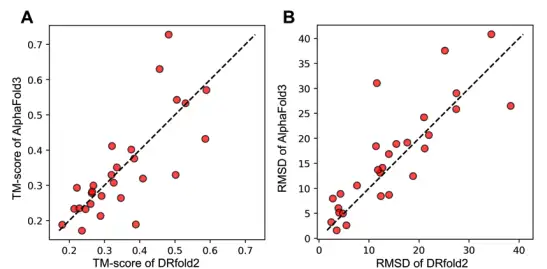
Um auf dieser Grundlage die Leistung von DRfold2 und AlphaFold3 bei der Vorhersage der RNA-3D-Struktur zu vergleichen, übermittelten die Forscher die RNA-Sequenzen im Testsatz auch an den AlphaFold-Server und erhielten die vorhergesagte Struktur von AlphaFold3 unter Verwendung der Standard-Seed-Konfiguration. sich herausstellen,Der durchschnittliche TM-Score (0,351) und RMSD (14,6 Å) von DRfold2 sind etwas höher als die von AlphaFold3 (0,345 und 16,0 Å).
Erwähnenswerter ist, dass DRfold2 und AlphaFold3 zwar eine ähnliche Gesamtleistung aufweisen, die Ergebnisse in der folgenden Abbildung jedoch die starke Komplementarität zwischen den beiden hervorheben, insbesondere wenn die Vorhersage erheblich von der Diagonalen abweicht.Durch die Einbeziehung der Vorhersagen von AlphaFold3 als zusätzlichen potenziellen Funktionsterm in das DRfold2-Optimierungsframework erzielten die Forscher statistisch signifikante Verbesserungen sowohl beim TM-Score als auch beim RMSD.
Das Team von Professor Zhang Yang konzentriert sich seit vielen Jahren auf die Forschung im Bereich KI und Computerbiologie
Das in dieser Studie vorgeschlagene DRfold2 ist eigentlich eine verbesserte Version des DRfold-Modells, das zuvor vom Team von Professor Zhang Yang vorgeschlagen wurde.
Im September 2023 veröffentlichte das Team von Professor Zhang Yang in der Zeitschrift Nature Communications einen Artikel mit dem Titel „Integrating end-to-end learning with deep geometrical potentials for ab initio RNA structure prediction“.
Diese Studie berichtet über eine neue Technologie, DRfold, zur genauen Vorhersage der dreidimensionalen Struktur von RNA.Die Kerninnovation liegt in der Einführung zweier komplementärer potentieller Energiefunktionen: FAPE-Potenzial und geometrisches Potenzial.Sie werden über zwei unabhängige Transformer-Netzwerke trainiert und stellen zusammen ein Deep-Learning-Potenzial für die Vorhersage von RNA-Strukturen dar. Rechenergebnisse zeigen, dass DRfold im Vergleich zu früheren Computermethoden zur Vorhersage der RNA-Struktur diese Methoden in vielen Leistungsindikatoren übertrifft.

Papieradresse:
https://www.nature.com/articles/s41467-023-41303-9
Von DRfold bis DRfold2 hat sich das Team von Professor Zhang Yang viele Jahre lang auf die Forschung im Bereich künstliche Intelligenz und Computerbiologie konzentriert. Sein Labor ist eines der ersten Labore, das auf Deep Machine Learning basierende Forschung zur Vorhersage von Protein- und RNA-Strukturen durchführt. Es wurden ihm Auszeichnungen wie der US-amerikanische Sloan Award, der US-amerikanische National Science Foundation Career Award und der University of Michigan Basic Science Research Award verliehen. Seit 2015 wurde es sieben Mal in die Liste der am häufigsten zitierten Wissenschaftler von Thomson Reuters/Clarivate Analytics aufgenommen. Der von seinem Labor entwickelte I-TASSER-Algorithmus (https://zhanggroup.org/I-TASSER/), Seit 2006 wurde es im weltweiten CASP-Experiment neun Mal in Folge als die genaueste Methode zur automatischen Vorhersage von Proteinstrukturen bewertet.
Am 2. Januar 2024 veröffentlichte das Team von Professor Zhang Yang in der Zeitschrift Nature Methods einen Artikel mit dem Titel „Verbesserung der Deep-Learning-Vorhersage von Proteinmonomeren und komplexen Strukturen durch Verwendung von DeepMSA2 mit riesigen Metagenomikdaten“.
Im Rahmen der Studie wurden zwei neue Softwareprogramme entwickelt, um die Genauigkeit der Strukturvorhersage von Proteininteraktionen zu verbessern. Die Autoren entwickelten DeepMSA2, das rekursive dynamische Programmierung und Hidden-Markov-Modellalgorithmen verwendet, um schnell hochwertige MSA-Daten aus riesigen metagenomischen Sequenzbibliotheken zu extrahieren, und dann die neu entwickelte DMFold-Software verwendet, um die dreidimensionale Struktur des Proteinkomplexes zu konstruieren.
Experimentelle Ergebnisse zeigen, dass die Genauigkeit der strukturellen Vorhersage von DMFold/DeepMSA2 für Proteinkomplexe deutlich besser ist als die von Algorithmen wie AlphaFold2. Insbesondere DMFold (https://zhanggroup.org/DMFold)-Algorithmus hat im letzten Wettbewerb zur Vorhersage von Proteinstrukturen (CASP15) die Meisterschaft in der Vorhersage von Proteinkomplexstrukturen gewonnen.

Papieradresse:
https://www.nature.com/articles/s41592-023-02130-4
Vor kurzem hat das Team seine Forschungsrichtung weiter ausgeweitet und sich nun auch mit dem Design und der Strukturvorhersage von RNA und kurzen Peptiden befasst. Darüber hinaus hat es sich mit Themen im Zusammenhang mit dem Arzneimitteldesign befasst. Ich bin überzeugt, dass Professor Zhang Yang auch in Zukunft sein Team anführen wird, um die Geheimnisse der Biologie zu erforschen.
Quellen:
1.https://www.biorxiv.org/content/10.1101/2025.03.05.641632v1
2.https://mp.weixin.qq.com/s/X_VJ-WOWEP08p5GAJOgq9A








