Command Palette
Search for a command to run...
AlphaFold Erreicht Einen Neuen Meilenstein in Der Anwendung! Team Der Universität Cambridge Schlägt AlphaFold-Metainferenz Zur Genauen Vorhersage Ungeordneter Proteinstruktursätze Vor
Seit dem Aufkommen von AlphaFold Ende 2018 hat sich das Feld der Proteinstrukturvorhersage mit Unterstützung der KI enorm verändert. Heute beeindruckt AlphaFold nicht nur durch seine Vorhersagegenauigkeit, sondern erweitert in der neuesten Iteration auch schrittweise seinen Vorhersagebereich. Kein Wunder also, dass Shi Yigong, ein Mitglied der Chinesischen Akademie der Wissenschaften, dies einmal vor den Medien großzügig kommentierte: „Meiner Meinung nach ist dies der größte Beitrag der künstlichen Intelligenz auf dem Gebiet der Wissenschaft. Es ist zugleich einer der wichtigsten wissenschaftlichen Durchbrüche der Menschheit im 21. Jahrhundert. Es ist eine ganz bemerkenswerte historische Errungenschaft in der wissenschaftlichen Erforschung der Natur durch die Menschheit.“
Obwohl die von AlphaFold angeführte Revolution in der Vorhersage von Proteinstrukturen so schnell voranschreitet, stehen noch immer einige ungelöste Probleme im Weg. Unter ihnen war die Erforschung ungeordneter Proteine schon immer ein schwieriges Problem im Bereich der Biowissenschaften. Diese Proteine spielen eine Schlüsselrolle bei der Zellsignalisierung, bei Regulierungsprozessen und einer Vielzahl von Krankheiten.Aufgrund ihrer besonderen Heterogenität und Strukturdynamik können sie jedoch nicht durch eine einzige Struktur dargestellt werden.Daher wurden in dieser Forschung keine so großen Fortschritte erzielt wie bei der Vorhersage geordneter Proteinstrukturen. Der Erfolg von AlphaFold hat Wissenschaftlern neue Wege zur Problemlösung aufgezeigt.
Kürzlich veröffentlichte ein Forschungsteam der Universität Cambridge eine neue Studie und schlug eine Methode namens AlphaFold-Metainference vor.Diese Methode verwendet die Korrelation zwischen der von AlphaFold vorhergesagten Karte des ausgerichteten Fehlers (PAE) und der Distanzänderungsmatrix in der molekularen Dynamiksimulation (MD), um strukturelle Ensembles ungeordneter Proteine und Proteine mit ungeordneten Regionen zu konstruieren.Es liefert neue Ideen für die Vorhersage ungeordneter Proteinstrukturen auf Basis von Deep-Learning-Methoden und erweitert zudem den Anwendungsbereich von AlphaFold.
Die entsprechenden Forschungsergebnisse wurden aktuell in der internationalen Fachzeitschrift Nature Communications unter dem Titel „AlphaFold prediction of structural ensembles of disordered proteins“ veröffentlicht.
Forschungshighlights:
* Durchbrechen Sie die Grenzen der Vorhersage und erreichen Sie hochpräzise Vorhersagen. Die Studie bestätigte, dass AlphaFold die Abstände zwischen Rückständen genau vorhersagen kann, auch ohne mit ungeordneten Proteindaten trainiert zu werden.
* Vorhersagemethoden erneuern und Struktursammlungen aufbauen. Diese Methode verwendet die von AlphaFold vorhergesagte Distanz als strukturelle Einschränkung und kombiniert das Meta-Inferenz-Framework und die molekulare Dynamiksimulation, um eine Sammlung von Strukturen ungeordneter Proteine und Proteine mit ungeordneten Regionen zu erstellen.
* Deep-Learning-Methoden vertiefen und Anwendungsgrenzen erweitern. Diese Methode eignet sich gut für den Umgang mit stark und teilweise ungeordneten Proteinen. Der generierte Struktursatz weist eine wesentlich höhere Übereinstimmung mit den experimentellen Daten auf als eine einzelne AlphaFold-Struktur, wodurch das Problem der Vorhersage ungeordneter Proteinstrukturen effektiv gelöst wird.

Papieradresse:
https://www.nature.com/articles/s41467-025-56572-9
Das Open-Source-Projekt „awesome-ai4s“ vereint mehr als 200 AI4S-Papierinterpretationen und stellt umfangreiche Datensätze und Tools bereit:
https://github.com/hyperai/awesome-ai4s
Datensatz: Strenge Überprüfung von Daten aus mehreren Quellen
Da es für das Training der Deep-Learning-Modelle nur sehr wenige Struktursammlungen ungeordneter Proteine gibt und diese auch nur sehr wenige genau sind, diese jedoch auf Grundlage der verfügbaren Informationen über geordnete Proteine vorhergesagt werden können, verwendeten die Forscher zum Trainieren der Deep-Learning-Modelle eine große Anzahl hochauflösender gefalteter Proteinstrukturen in der Protein Data Bank (PDB).
Im Hinblick auf den Vergleich experimenteller Daten ist es eine Herausforderung, experimentelle Informationen über die Abstände zwischen Resten in ungeordneten Proteinen zu erhalten, und die Datenbeschriftungen selbst können die Eigenschaften des Konformationsensembles beeinflussen.Dazu nutzten die Forscher Daten der Kleinwinkel-Röntgenstreuung (SAXS) und Diffusionsmessungen mittels Kernspinresonanz (NMR).Es liefert markierungsfreie Informationen über die Abstandsverteilung zwischen ungeordneten Proteinresten für die Forschung, die zum Vergleich und zur Überprüfung der Vorhersageergebnisse verwendet werden.
Darüber hinaus, in weiterer Überprüfung,Die Forscher analysierten außerdem die Strukturensembledaten von Aβ und α-Synuclein, die sie durch All-Atom-Moleküldynamiksimulationen und grobkörnige Simulationen mit CALVADOS-2 (C2) gewonnen hatten.Dies bestätigt weiter die Genauigkeit der von AlphaFold vorhergesagten Entfernung.
Modellarchitektur: Innovative Fusion-Meta-Reasoning-Methode
Die in dieser Studie beschriebene AlphaFold-Metainference-Methode wird verwendet, um eine Sammlung von Strukturen zu generieren, die die nativen Zustände ungeordneter Proteine und Proteine mit ungeordneten Regionen darstellen.
Der Kern des Ansatzes basiert auf der Beobachtung, dass die von AlphaFold vorhergesagten Abstände zwischen den Aminosäureresten selbst für ungeordnete Proteine relativ genau sind und daher als strukturelle Einschränkungen in molekulardynamischen Simulationen innerhalb eines Meta-Inferenz-Rahmens verwendet werden können. Einfach ausgedrückt verwendet AlphaFold-Metainference zur Generierung des Strukturensembles die vorhergesagten Abstände als strukturelle Einschränkungen in der molekulardynamischen Simulation.Konvertieren Sie AlphaFold-Distanzkarten (Distogramme) in Struktursätze.
Zuerst kommt die AlphaFold-Vorhersagedistanz. Die Forscher verwendeten die Distanzkarte von AlphaFold, um die durchschnittliche Distanz zwischen Rückständen vorherzusagen, und berechneten die vorhergesagte Distanz und Standardabweichung mithilfe einer bestimmten Formel. Anschließend wurde eine Mehrfachsequenzausrichtung basierend auf MMseqs2 durchgeführt und die Vorhersage mithilfe des AlphaFold 1.1.1-Modells mit Standardeinstellungen und ohne Verwendung einer Strukturvorlage durchgeführt. Die von AlphaFold ausgegebenen Abstände zwischen den Resten sind auf 64 gleich breite Bins verteilt, die von 2,15625 bis 21,84375 Å reichen, wobei der letzte Bin auch Abstände über 21,84375 Å enthält.
Anschließend wird die Meta-Reasoning-Methode kombiniert. Das sogenannte Meta-Reasoning ist eine Bayes'sche Denkmethode, die durch die Kombination von Vorinformationen und experimentellen Daten auf Basis des Maximum-Entropie-Prinzips den Struktursatz bestimmen kann. In diesem StadiumDie Forscher verwendeten den von AlphaFold vorhergesagten Distanzgraphen als pseudo-experimentelle Daten und wandten die Bayessche Metainferenzmethode an.Bestimmen Sie das strukturelle Ensemble, indem Sie strukturelle Heterogenität von systematischen Fehlern trennen, wie etwa Ungenauigkeiten im Kraftfeld oder Vorwärtsmodell, zufälligen Fehlern in den Daten und Fehlern aufgrund der begrenzten Stichprobengröße im Ensemble.
Bei molekulardynamischen Simulationen werden Berechnungen auf Grundlage der Meta-Inferenz-Energiefunktion durchgeführt und Fehlerparameter durch mehrere Replikationssimulationen und Gibbs-Stichproben ermittelt.Schließlich wurde das Kraftfeld von CALVADOS-2 verwendet, um eine grobkörnige Simulation durchzuführen.Implementieren Sie AlphaFold-Metainferenz.
Der letzte Schritt ist die Auswahl der Distanzbeschränkung. In dieser Phase wird die von AlphaFold vorhergesagte Distanz basierend auf der Distanzwahrscheinlichkeit und dem vorhergesagten Ausrichtungsfehler gefiltert.Die Auswahlkriterien wurden durch die Kombination von Proteinhydrophilie und vorhergesagten Werten des Local Distance Difference Test (pLDDT) bestimmt.Es ist erwähnenswert, dass die experimentelle Verwendung von pLDDT-Scores zur Auswahl von Restabständen in strukturierten Regionen ihre Verwendung als Abstandsbeschränkungen zur Optimierung der Generierung von Strukturensembles nicht ausschließt.
Alle molekulardynamischen Simulationen gingen von der von AlphaFold vorhergesagten Struktur aus und wurden unter dem NVT-Ensemble durchgeführt. Für jede Simulation wurden sechs Replikate eingerichtet, jedes Replikat umfasste 1 Million Schritte und die Simulation startete von unterschiedlichen Anfangspositionen, die im Schritt der Energieminimierung ermittelt wurden.Die Simulation verwendet einen Langevin-Integrator.Der Zeitschritt beträgt 5 fs, der Reibungskoeffizient beträgt 0,01 ps⁻¹ und es wird ein Cα-basiertes Modell mit CALVADOS-2-Parametern und Funktionsform verwendet.
Unter anderem wurde für stark und teilweise ungeordnete Proteine PULCHRA verwendet, um alle Strukturen in der grobkörnigen Sammlung in Allatomdarstellungen umzuwandeln, und dann wurde GROMACS zur Energieminimierung verwendet, um genauere Strukturen zu erhalten.
Insgesamt veranschaulichen die von den Forschern vorgestellten Ergebnisse, wie Deep-Learning-Methoden, die ursprünglich zur Vorhersage des nativen Zustands gefalteter Proteine entwickelt wurden, genutzt werden können, um eine Sammlung von Strukturen zu generieren, die den nativen Zustand ungeordneter Proteine darstellen. Diese Methode erweitert den Umfang der auf Deep Learning basierenden Proteinstrukturvorhersage erheblich und bietet eine neue Idee für die Vorhersage ungeordneter Proteinstrukturen.
Experimentelle Ergebnisse: vollständig überprüfen seine Rationalität
In Bezug auf die AlphaFold-Vorhersagegenauigkeit
Die Forscher verglichen eine Gruppe von 11 Proteinen, für die sowohl SAXS- als auch NMR-Diffusionsmessungen verfügbar waren, und stellten eine gute Übereinstimmung zwischen den von AlphaFold vorhergesagten Distanzverteilungen und den aus SAXS abgeleiteten Distanzverteilungen fest. Die Forscher fügten außerdem ein gefaltetes Protein als Kontrolle hinzu, wie in der Abbildung unten gezeigt.
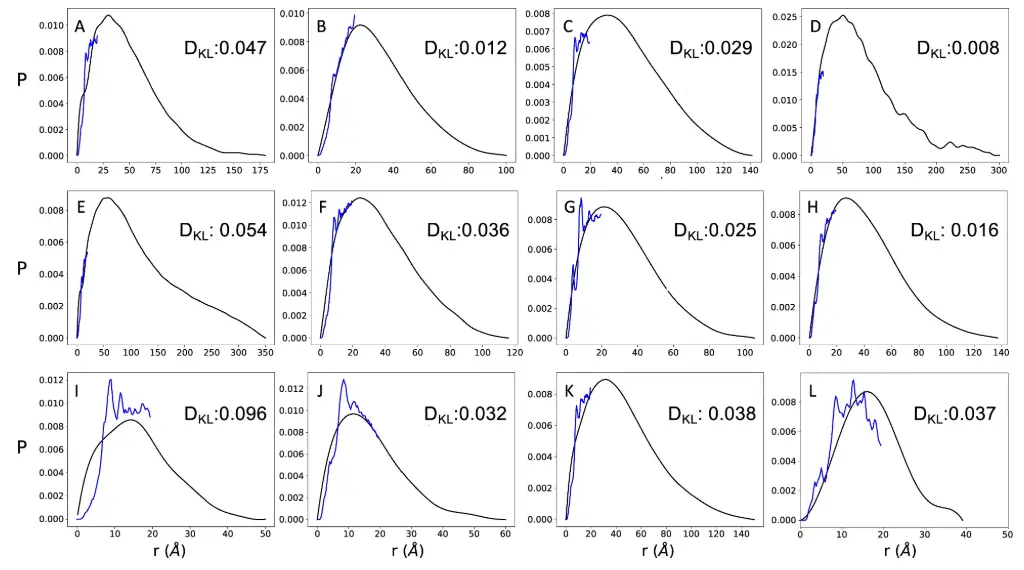
Es ist erwähnenswert, dass die von AlphaFold vorhergesagte Distanzverteilung nicht die gesamte von SAXA abgeleitete Verteilung abdeckt, da die von AlphaFold vorhergesagte Distanz bis zu etwa 22 Å beträgt. Die Ergebnisse zeigten, dass der DKL-Wert der hinzugefügten Kontrollgruppe 0,037 betrug, was mit den DKL-Werten von 11 stark ungeordneten Proteinen vergleichbar war (DKL-Bereich lag bei 0,008–0,096).Dies zeigt weiter, dass AlphaFold eine vergleichbare Genauigkeit bei der Vorhersage von Abständen zwischen Resten für ungeordnete und geordnete Proteine aufweist.
Darüber hinaus stimmen die von AlphaFold vorhergesagten Abstände auch gut mit den aus den MD-Ensembles von Aβ und α-Synuclein und aus dem CALVADOS-2-Ensemble zurückberechneten Abständen überein.
Bei der Verifikation hochgradig ungeordneter Struktursammlungen
Die paarweise Abstandsverteilung kann mithilfe von Kleinwinkel-Röntgenstreuungsmessungen berechnet werden. Die Forscher verglichen die experimentell erhaltene Distanzverteilung mit der Distanzverteilung, die sich aus dem Satz von Strukturen ergab, die durch AlphaFold-Metainference-Simulationen ermittelt wurden, wiederum für die oben erwähnten elf stark ungeordneten Proteine.
Gleichzeitig zeigten die Forscher zum weiteren Vergleich auch die mit CALVADOS-2 erhaltene Distanzverteilung sowie die von AlphaFold abgeleitete Distanzverteilung, die direkt aus einer einzelnen AlphaFold-Struktur generiert wurde. Um einen quantitativen Vergleich zu ermöglichen, stellten die Forscher fest, dass das Ensemble der Strukturen, das durch AlphaFold-Metainference mit CALVADOS-2 bereitgestellt wurde, mit den SAXS-Daten konsistenter war als eine einzelne aus AlphaFold abgeleitete Struktur.
Die Forscher verglichen die Strukturensembles außerdem mithilfe von chemischen Verschiebungen in der NMR-Spektroskopie, die für jeden Zeitschritt mithilfe von CamShift zurückgerechnet wurden.Die Ergebnisse zeigen, dass die Vorhersagen von AlphaFold-Metainference in einigen Fällen genauer sind.Wie in der Abbildung unten gezeigt.
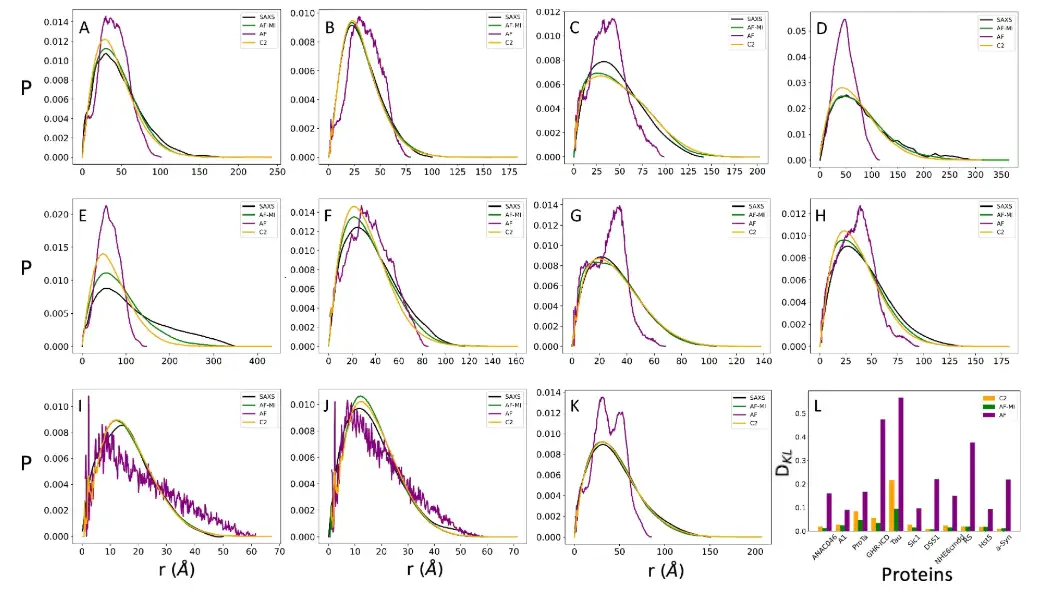
* Die Verteilung der experimentellen paarweisen Abstände, die durch SAXS ermittelt wurden, wird als schwarze Linie dargestellt
* Die Vorhersage einzelner AlphaFold-Strukturen wird durch eine violette Linie dargestellt
* AlphaFold-Metainference-Strukturensemblevorhersagen werden als grüne Linien dargestellt
* Die von CALVADOS-2 ermittelte paarweise Distanzverteilung wird als orange Linie dargestellt
Bei der Überprüfung teilweise ungeordneter strukturierter Sammlungen
Die Forscher bereiteten einen Satz von sechs Proteinen mit sowohl geordneten als auch ungeordneten Domänen und unterschiedlichen Sequenzlängen vor, für die SAXS-Daten zur Überprüfung verfügbar waren.
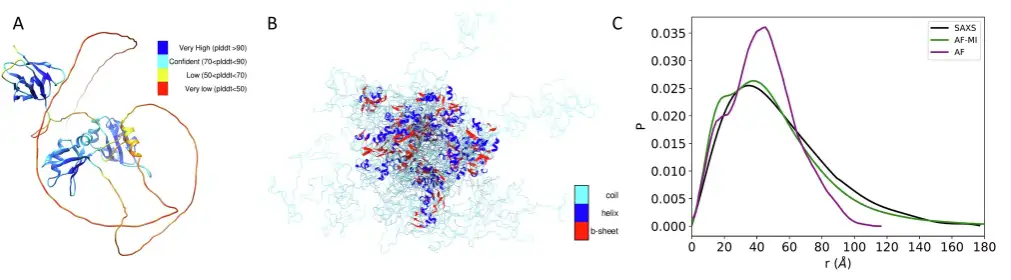
Das erste ist TDP-43, ein multifunktionales RNA-bindendes Protein mit modularer Struktur, das an einer Vielzahl von zellulären Prozessen beteiligt ist, darunter Transkription, Prä-mRNA-Spleißen und Regulierung der mRNA-Stabilität, und das mit ALS und anderen neurodegenerativen Erkrankungen in Verbindung gebracht wird.
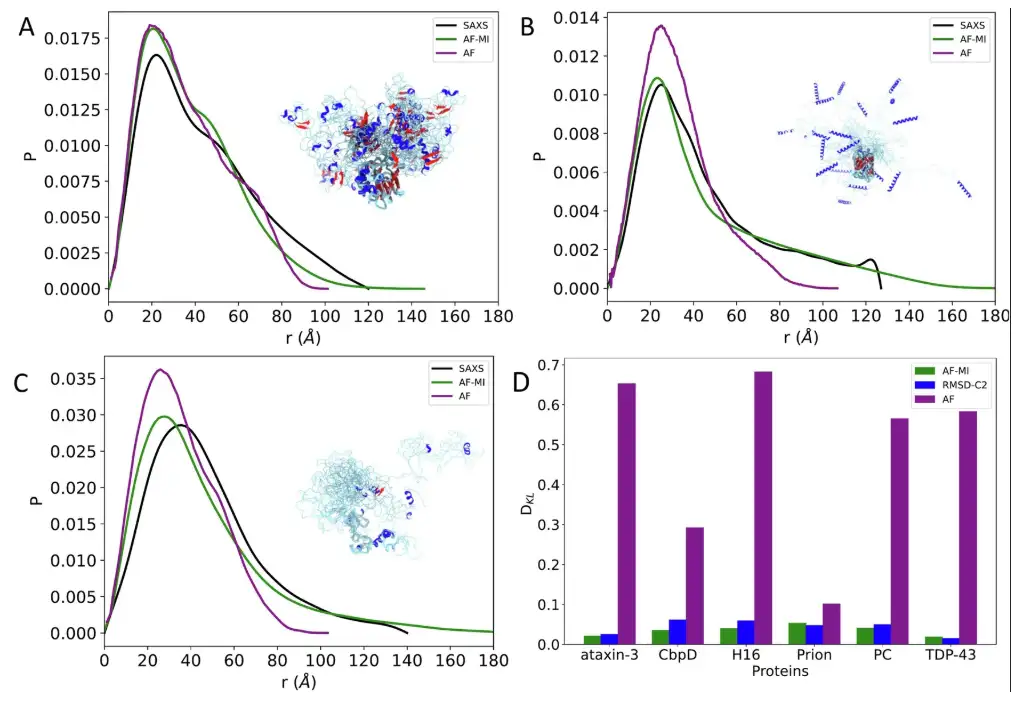
Die experimentellen Ergebnisse zeigten, dass bei der Anwendung der Filterkriterien der Forscher zur Auswahl der von AlphaFold vorhergesagten Distanzen und der anschließenden Anwendung von AlphaFold-Metainference mit diesen Distanzbeschränkungen,Das erhaltene Strukturensemble steht in deutlich besserer Übereinstimmung mit den SAXS-Daten.Der DKL-Wert beträgt nur 0,018.Dies ist besser als der DKL-Wert von 0,582 bei Verwendung der von AlphaFold vorhergesagten Struktur direkt mit SAXS-Daten.Wie in der Abbildung unten gezeigt.
Anschließend analysierten die Forscher Ataxin-3 und menschliches Prionprotein. Für Ersteres wurden ähnliche Ergebnisse wie für TDP-43 erhalten, wie oben beschrieben. Hier war die mit AlphaFold direkt aus der AlphaFold-Proteinstrukturdatenbank ermittelte Struktur schlecht mit den SAXS-Daten vereinbar (DKL-Wert 0,653). Bei Anwendung eines Filterkriteriums zur Auswahl der von AlphaFold aus der AlphaFold-Metainference-Simulation vorhergesagten Distanzen hingegenEs wurde eine Reihe von Strukturen erhalten, die besser mit den SAXS-Daten übereinstimmen.Der DKL-Wert beträgt nur 0,020. Wie in der Abbildung unten gezeigt.

Für Letzteres ist die vorhergesagte Struktur, die direkt aus der AlphaFold-Proteinstrukturdatenbank mithilfe von AlphaFold gewonnen wurde, schlecht konsistent mit den SAXS-Daten, mit einem DKL-Wert von 0,1,Durch die Anwendung von Filterkriterien wurde ein Satz von Strukturen erhalten, der besser mit den SAXS-Daten übereinstimmte.Der DKL-Wert beträgt nur 0,053. Wie in der Abbildung unten gezeigt.
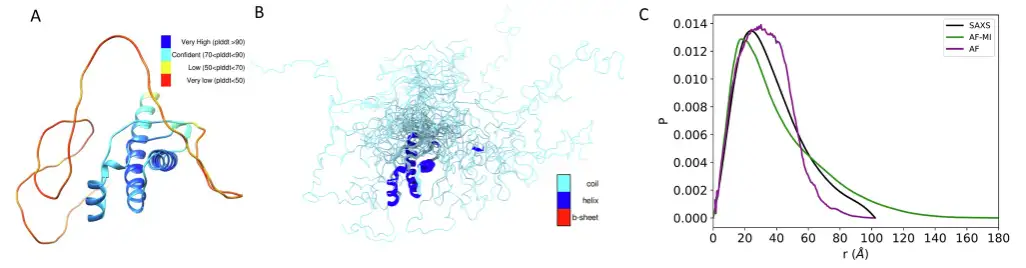
Darüber hinaus untersuchten die Forscher drei weitere Proteine: CbpD, H16 und PC. Die Ergebnisse zeigten, dassIn allen Fällen ist die Übereinstimmung zwischen der experimentellen und der rückberechneten Inter-Rest-Abstandsverteilung sehr gut.Und es stellt eine erhebliche Verbesserung gegenüber der AlphaFold-Einzelstruktur dar, die direkt aus der AlphaFold-Proteinstrukturdatenbank gewonnen wurde, wie in Abbildung D unten dargestellt.
Schließlich zeigte AlphaFold-Metainference im Vergleich zur CALVADOS-2-Methode bei vier der sechs Proteine (Ataxin-3, CbpD, H16 und PC) eine bessere Leistung und erzeugte bei den verbleibenden zwei (TDP-43 und menschliches Prionprotein) vergleichbare Strukturensembles. Wie in der Abbildung unten gezeigt.
Fortschritte bei der Vorhersage ungeordneter Proteine auf Basis von Deep Learning
In den letzten Jahren wurde AlphaFold hauptsächlich zur Vorhersage der statischen Struktur gefalteter Proteine verwendet, was ihm auch Kritik seitens der wissenschaftlichen Forschungsgemeinschaft einbrachte. Diese Studie bestätigt zweifellos, dass sie auch potenzielle Anwendungsvorteile bei der Vorhersage ungeordneter Proteinstrukturen bietet und bietet zudem eine neue Forschungsrichtung für die Vorhersage ungeordneter Proteinstrukturen.
Tatsächlich ist es durch die enge Integration von KI und BiowissenschaftenEs gab viele Diskussionen über die Vorhersage ungeordneter Proteinstrukturen.Auch im Bereich der modernen Biowissenschaften ist der Einsatz von KI zur Aufklärung der Geheimnisse des Lebens zu einer gängigen Methode geworden.
So wurde beispielsweise in einem zuvor in Current Opinion in Structural Biology veröffentlichten Artikel der Anwendungsfortschritt von Deep Learning bei der Erforschung intrinsisch ungeordneter Proteine (IDPs) erörtert und seine Rolle bei der Förderung der Vorhersage ungeordneter Proteine und der Charakterisierung konformationeller Ensembles erläutert.
Die entsprechende Forschung wurde unter dem Titel „Deep Learning für intrinsisch ungeordnete Proteine: Von verbesserten Vorhersagen zur Entschlüsselung von Konformationsensembles“ veröffentlicht.
* Papieradresse:
https://www.sciencedirect.com/science/article/pii/S0959440X24001775
Zufällig veröffentlichte ein Forscherteam der Universität Kopenhagen in Dänemark in Nature einen Artikel über die Erforschung ungeordneter Proteine mit dem Titel „Conformational ensembles of the human intrinsically disordered proteome“. Der Artikel befasst sich mit der Verwendung verschiedener Deep-Learning-Methoden zur Vorhersage ungeordneter Regionen, konformationeller Ensembles und verwandter Eigenschaften von IDPs, darunter Deep-Learning-Methoden wie das oben erwähnte AlphaFold sowie Proteinsprachenmodelle, generative kontradiktorische Netzwerke usw.
*Papieradresse:
https://www.nature.com/articles/s41586-023-07004-5
Es besteht kein Zweifel, dass die rasante Entwicklung der KI unser Verständnis vom wahren Sinn des Lebens beschleunigt. Der britische Wissenschaftler John Kendrew brauchte einst 12 Jahre, um mithilfe der Röntgenkristallographie die erste Proteinstruktur zu erforschen. Jetzt braucht AlphaFold nur noch wenige Jahre, um das Geheimnis der Faltung von Hunderten Millionen Proteinen zu lüften. Wer kann in Zukunft behaupten, dass uns die Vorhersage ungeordneter Proteinstrukturen nicht gelingt?








