Command Palette
Search for a command to run...
Ausgewählt Für AAAI 2025! Die Tsinghua University/UCL Entwickelte Eine Lösung Zur Fusion Von Protein-RNA-Sprachmodellen, Die Affinitätsvorhersage Mit Der Aktualisierung Von SOTA Kombinierte
Alzheimer-Krankheit, Parkinson-Krankheit, Epilepsie ... Diese „notorisch furchterregenden“ neurodegenerativen Erkrankungen sind unsichtbare Killer für die Gesundheit älterer Menschen, und das Auftreten dieser Krankheiten hängt oft mit der abnormalen Bindung zwischen Protein und RNA zusammen.
Im biomedizinischen Bereich ist die Untersuchung der Protein-RNA-Bindung von entscheidender Bedeutung, da sie eine zentrale Rolle bei zahlreichen biologischen Prozessen spielt, etwa bei der Regulierung der Genexpression, der RNA-Verarbeitung und -Spleißen, der Translationsregulierung und der zellulären Stressreaktion.Das Verständnis des Mechanismus der Protein-RNA-Bindung ist der Schlüssel zur Aufdeckung komplexer Genregulationsprozesse und zur Analyse der genetischen Grundlagen von Krankheiten. Gleichzeitig finden Protein-RNA-Interaktionen auch in der gezielten RNA-Therapie wichtige Anwendung und eröffnen neue Wege für die Behandlung von Krebs, genetischen Erkrankungen und Viruserkrankungen.
Zu den ausgewählten Errungenschaften, die kürzlich auf der 39. jährlichen AAAI-Konferenz für Künstliche Intelligenz (AAAI 2025), der wichtigsten internationalen Konferenz für Künstliche Intelligenz, bekannt gegeben wurden,Ein gemeinsames Team der Tsinghua-Universität, des University College London, der Monash University und der Beijing University of Posts and TelecommunicationsvorschlagenDas CoPRA-Modell hat in der Branche große Aufmerksamkeit erregt und wurde für die mündliche Prüfung ausgewählt.
Dies ist der erste Versuch, das Protein-Sprachmodell (PLM) und das RNA-Sprachmodell (RLM) durch eine komplexe Strukturarchitektur zur Vorhersage der Protein-RNA-Bindungsaffinität zu kombinieren.Um die Leistung von CoPRA zu testen, stellten die Forscher den größten Datensatz zur Protein-RNA-Bindungsaffinität aus mehreren Datenquellen zusammen und bewerteten die Modellleistung anhand von drei Datensätzen. Die Ergebnisse zeigten, dass CoPRA bei mehreren Datensätzen eine hochmoderne Leistung erreichte.
Die zugehörigen Ergebnisse tragen den Titel „CoPRA: Bridging Cross-domain Pretrained Sequence Models with Complex Structures for Protein-RNA Binding Affinity Prediction“ und wurden als Vorabdruck auf arXiv veröffentlicht.

Papieradresse:
https://arxiv.org/abs/2409.03773
CoPRA-Lageradresse:
https://github.com/hanrthu/CoPRA
Das Open-Source-Projekt „awesome-ai4s“ vereint mehr als 200 AI4S-Papierinterpretationen und stellt umfangreiche Datensätze und Tools bereit:
https://github.com/hyperai/awesome-ai4s
Die biomedizinische Forschung treibt die Protein-RNA-Interaktionen weiter voran
In den vergangenen Jahren haben Forscher im biomedizinischen Bereich nie aufgehört, Protein-RNA-Interaktionen zu untersuchen und dabei erhebliche Fortschritte erzielt.
Die CLIP-Experimentaltechnologie ist eine der wichtigsten Technologien in der RNA-Forschung. Es kann die Bindungskarte des RNA-Bindungsproteins (RBP) im gesamten Transkriptom analysieren und ist die Grundlage für das systematische Verständnis der Funktion eines RBP und seines Regulationsmechanismus. CLIP-Experimente sind jedoch zeit- und arbeitsintensiv, können jeweils nur die RNA-Bindungsstelle eines bestimmten RBP in einer bestimmten Zellumgebung bereitstellen und stellen hohe Anforderungen an das experimentelle Material. Allerdings kann sich die Bindung von Proteinen und RNA bei Veränderungen in der Zellumgebung stark ändern. Für die Untersuchung der Regulierung von Proteinen auf RNA sind jedoch Bindungsinformationen in derselben Zellumgebung erforderlich.
Um das Problem der dynamischen Veränderungen der RBP-Bindung in verschiedenen zellulären Umgebungen zu lösen,Im Februar 2021 veröffentlichte die Forschungsgruppe von Zhang Qiangfeng am Center for Advanced Innovation in Structural Biology der Tsinghua-Universität in der Zeitschrift Cell Research ein Forschungsergebnis mit dem Titel „Vorhersage dynamischer zellulärer Protein-RNA-Interaktionen durch Deep Learning unter Verwendung von In-vivo-RNA-Strukturen“. In dieser Arbeit wurde das icSHAPE-Experiment verwendet, um die RNA-Sekundärstrukturkarten von sieben gängigen Zelltypen zu analysieren. Außerdem wurde ein künstlicher Intelligenzalgorithmus entwickelt, um die aus dem Experiment erhaltene intrazelluläre RNA-Struktur und die RBP-Bindungsinformationen der entsprechenden Zellumgebung zu integrieren. Zudem wurde eine neue PrismNet-Methode zur Vorhersage der dynamischen Bindung von intrazellulärem RBP auf der Grundlage von Informationen zur intrazellulären RNA-Struktur etabliert.
Um die Protein-RNA-Bindungsaffinität vorherzusagen, wurden in der Industrie mehrere rechnergestützte Methoden vorgeschlagen.Beinhaltet sequenzbasierte und strukturbasierte Methoden. Sequenzbasierte Methoden verarbeiten Protein- und RNA-Sequenzen separat mithilfe unterschiedlicher Sequenz-Encoder und modellieren anschließend die Interaktionen zwischen ihnen. Allerdings ist die Leistung dieser Ansätze oft begrenzt, da die Bindungsaffinität in erster Linie durch die Struktur der Bindungsschnittstelle bestimmt wird. Andere kürzlich vorgeschlagene Methoden konzentrieren sich auf die Extraktion struktureller Merkmale der Bindungsschnittstelle, wie Energie und Kontaktabstand. Basierend auf diesen extrahierten Merkmalen entwickelten die Forscher einen strukturbasierten maschinellen Lernansatz zur Affinitätsvorhersage. Aufgrund der begrenzten Datensatzgröße sind diese Methoden jedoch nur begrenzt auf neue Stichproben verallgemeinerbar und stark von der Merkmalsentwicklung abhängig.
Mit dem Aufkommen der künstlichen Intelligenz wurden viele Protein-Sprachmodelle (PLMs) und RNA-Sprachmodelle (RLMs) entwickelt, die bei verschiedenen nachgelagerten Aufgaben hervorragende Leistung und Generalisierungsfähigkeiten bewiesen haben.Da die dreidimensionale Struktur von Proteinen/RNAs für das Verständnis ihrer Funktionen von entscheidender Bedeutung ist, ist gleichzeitig die Einbeziehung struktureller Informationen in Sprachmodelle zu einem neuen Trend geworden.
Ein Team der University of Missouri, der University of Kentucky und der University of Alabama nutzte beispielsweise eine multiperspektivische Vergleichslerntechnologie, um wichtige Informationen zur Proteinstruktur in das Proteinsprachenmodell zu integrieren. Basierend auf diesem Konzept entwickelte das Team S-PLM: ein Proteinsprachenmodell mit der Fähigkeit, 3D-Strukturinformationen von Proteinen wahrzunehmen. S-PLM zeigt bei mehreren Aufgaben zur Proteinvorhersage eine hervorragende Leistung. Nach dem Training mit einem leichtgewichtigen Tuning-Tool erreicht oder übertrifft die Leistung von S-PLM die aktuellen State-of-the-Art-Methoden bei Aufgaben wie der Vorhersage von Proteinfunktionen, der Vorhersage von Enzymreaktionsklassen und der Vorhersage von Sekundärstrukturen. Die entsprechende Forschung wurde auf bioRxiv unter dem Titel „S-PLM: Structure-aware Protein Language Model via Contrastive Learning between Sequence and Structure“ veröffentlicht.
Obwohl aktuelle Branchenforschungen das große Potenzial biologischer Sprachmodelle auf Basis struktureller Informationen bei interaktiven Aufgaben aufgezeigt haben, sind Arbeiten, bei denen vorab trainierte Modelle aus unterschiedlichen biologischen Bereichen kombiniert werden, noch immer selten.In CoPRA, das gemeinsam von der Tsinghua-Universität, dem University College London, der Monash-Universität und der Beijing University of Posts and Telecommunications vorgeschlagen wurde, wurde erstmals der Versuch unternommen, Protein- und RNA-Sprachmodelle mit komplexen Strukturinformationen zu kombinieren, um die Protein-RNA-Bindungsaffinität vorherzusagen.
Entwurf eines leichten Co-Former-Modells zum Aufbau von CoPRA
Insgesamt ist der Konstruktionsprozess des CoPRA-Modells in der folgenden Abbildung dargestellt:
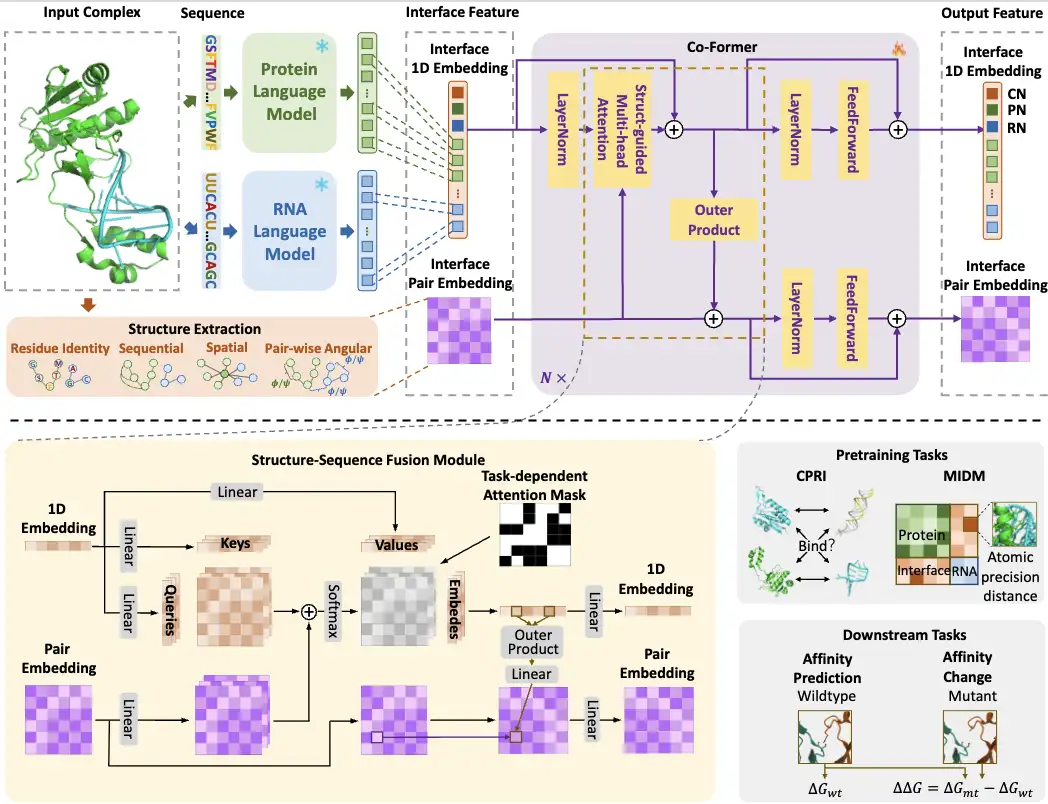
Erste,Die Forscher gaben Protein- und RNA-Sequenzen in PLM bzw. RLM ein und wählten dann die Einbettungen an der Interaktionsschnittstelle aus den Ausgaben der beiden Sprachmodelle als Sequenzeinbettungen für das anschließende kreuzmodale Lernen aus. Gleichzeitig werden auch Strukturinformationen (Schnittstellenmerkmale) aus der Interaktionsschnittstelle als gepaarte Einbettung extrahiert.
Dann,Die Forscher entwarfen ein leichtgewichtiges Co-Former-Modell, das Schnittstellensequenz-Einbettungen aus zwei Sprachmodellen mit komplexen Strukturinformationen kombiniert, um ein Struktur-Sequenz-Fusionsmodul zu bilden. Insbesondere verschmilzt Co-Former 1D- und paarweise Einbettungen über strukturgesteuerte Multi-Head-Self-Attention- und Outer-Product-Module und wendet aufgabenabhängige Aufmerksamkeitsmasken an. Die Ausgabe-Spezialknoten und gepaarten Einbettungen von Co-Former werden entsprechend verschiedener Aufgaben verwendet, darunter zwei Vortrainingsaufgaben und zwei nachgelagerte Affinitätsaufgaben.
Die Forscher schlugen außerdem eine Dual-Range-Vortrainingsstrategie für Co-Former vor.Zur Modellierung einer grobkörnigen kontrastiven Interaktionsklassifizierung (CPRI) und einer feinkörnigen Schnittstellendistanzvorhersage (MIDM), gelernt mit atomarer Genauigkeit.
Um die Leistung von CoPRA und anderen Modellen zu bewerten,Forscher müssen sich mit dem Mangel an einheitlichen Standarddatensätzen für Annotationen befassen. Sie sammelten also Proben aus drei öffentlichen Datensätzen: PDBbind, PRBABv2 und ProNAB, stellten den größten Datensatz zur Protein-RNA-Bindungsaffinität PRA310 zusammen und bewerteten die Fähigkeit ihres Modells, die Protein-RNA-Bindungsaffinität anhand der Datensätze PRA310 und PRA201 vorherzusagen.
*PRA201-Datensatz: eine Teilmenge von PRA310, jeder Komplex enthält nur eine Proteinkette und eine RNA-Kette und unterliegt strengeren Längenbeschränkungen
CoPRA liefert die beste Leistung bei der Vorhersage der Protein-RNA-Bindungsaffinität
Wie in der folgenden Tabelle gezeigt, erzielt die neu trainierte Version von CoPRA die beste Leistung beim PRA310-Datensatz. Darüber hinaus übertreffen die meisten Methoden, die LM-Einbettungen als Eingabe verwenden, andere Methoden, was auf das große Potenzial der Kombination vorab trainierter unimodaler LMs für die Affinitätsvorhersage hinweist.
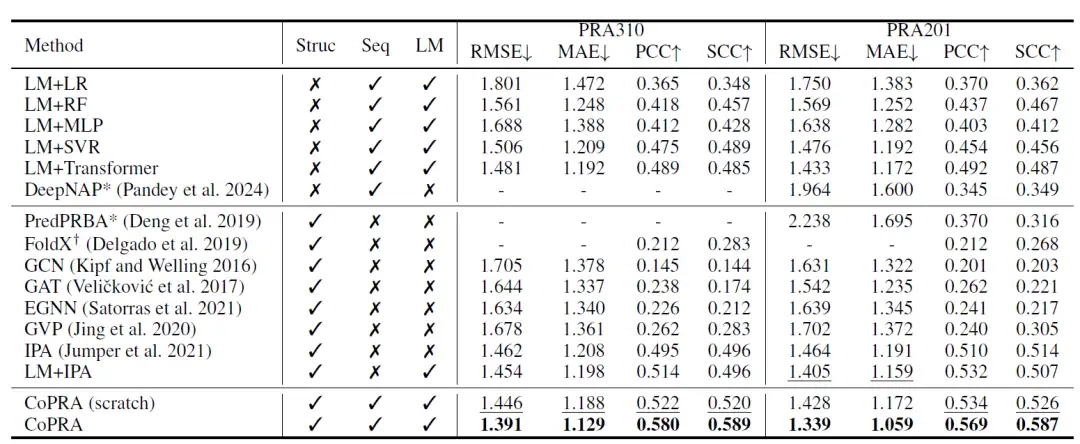
Anschließend trainierten die Forscher das Modell mithilfe ihres kompilierten, unüberwachten Datensatzes PRI30k vor und verbesserten so die Gesamtleistung bei beiden Datensätzen erheblich. Im PRA310-Datensatz erreicht CoPRA einen RMSE von 1,391, einen MAE von 1,129, einen PCC von 0,580 und einen SCC von 0,589, was viel besser ist als das zweitbeste Modell CoPRA (von Grund auf trainiert). PredPRBA und DeepNAP unterstützen die Vorhersage der Protein-RNA-Paaraffinität. Die Forscher verglichen die Leistung dieser Methoden mit dem PRA201-Datensatz und zeigten, dass ihre Leistung bei PRA201 deutlich niedriger war als ihre gemeldeten Ergebnisse, obwohl in ihren Trainingssätzen mindestens 100 Proben in PRA201 auftauchten, was darauf hindeutet, dass diese Methoden nur über geringe Generalisierungsfähigkeiten verfügen.
CoPRA ist besser in der Vorhersage der Auswirkungen von Mutationen auf die Bindungsaffinität und verfügt über eine ausgezeichnete Generalisierungsfähigkeit
Um das detaillierte Verständnis des Modells zur Affinität weiter zu evaluieren, haben die Forscher das Modell umgeleitet, um die Auswirkungen einzelner Punktmutationen im Protein auf den Protein-RNA-Komplex vorherzusagen. Unter Bezugnahme auf verwandte Studien zur Vorhersage von Proteinmutationseffekten haben die Forscher die Messwerte auf jeder komplexen Ebene gemittelt und die Zero-Shot- und Feinabstimmungsleistung von CoPRA nach dem Vortraining auf PRI30k und der Abstimmung auf PRA310 bewertet.
Wie in der folgenden Tabelle gezeigt, übertraf das in dieser Studie vorgeschlagene Modell nach der Feinabstimmung mithilfe des Kreuzvalidierungssatzes von mCSM andere Modelle in allen vier Indikatoren, mit einem RMSE von 0,957, einem MAE von 0,833, einem PCC von 0,550 und einem SCC von 0,570.
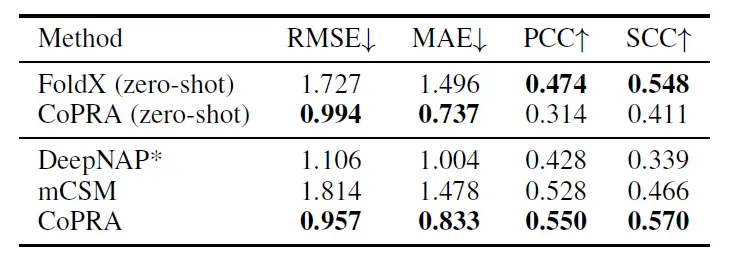
Diese überlegene Leistung ist auf das duale Vortrainingsziel zurückzuführen, obwohl keine mutierten komplexen Strukturen beobachtet wurden, und demonstriert die Generalisierungsfähigkeit von CoPRA bei verschiedenen affinitätsbezogenen Aufgaben.
Bahnbrechende Fortschritte bei multimodalen Proteinsprachenmodellen
Der Kern der oben vorgestellten Forschungsidee besteht darin, mehrere biologische Modalitäten wie Proteine und RNA mit komplexen Strukturinformationen zu kombinieren, was als sogenanntes multimodales Lernen bezeichnet wird. Einfach ausgedrückt ist multimodales Lernen der Prozess der Integration verschiedener Datentypen in ein Modell im Rahmen des Deep Learning.
In den letzten Jahren haben Forscher mit der rasanten Entwicklung großer Sprachmodelle begonnen, diese auf dem Gebiet der Proteinwissenschaft anzuwenden, um die Funktion, Struktur und Eigenschaften von Proteinen genau zu verstehen und vorherzusagen. Bisherige proteinorientierte Großsprachenmodelle verarbeiten jedoch hauptsächlich Aminosäuresequenzen als Text und können die umfangreichen Strukturinformationen von Proteinen nicht vollständig nutzen.Heute liefern die Fortschritte im multimodalen Lernen neue Ideen für immer mehr damit verbundene Forschung.
Im Bereich der Arzneimittelforschung und -entwicklung ist beispielsweise eine genaue und effektive Vorhersage der Bindungsaffinität zwischen Proteinen und Liganden für das Screening und die Optimierung von Arzneimitteln von entscheidender Bedeutung. Allerdings wurde in früheren Studien die wichtige Rolle molekularer Oberflächeninformationen bei Protein-Liganden-Interaktionen nicht berücksichtigt. Auf dieser GrundlageForscher der Universität Xiamen haben ein neuartiges Framework zur multimodalen Merkmalsextraktion (MFE) vorgeschlagen.Dieses Framework kombiniert erstmals Informationen zur Proteinoberfläche, 3D-Struktur und Sequenz und verwendet einen Cross-Attention-Mechanismus, um Merkmale zwischen verschiedenen Modalitäten auszurichten. Experimentelle Ergebnisse zeigen, dass diese Methode bei der Vorhersage der Protein-Liganden-Bindungsaffinität Spitzenleistungen erzielt. Die entsprechende Forschung wurde im Juni 2024 in Bioinformatics unter dem Titel „Surface-based multimodal protein–ligand binding affinity prediction“ veröffentlicht.
Im Dezember 2024 schlug ein Forschungsteam der East China Normal University und anderer Institutionen eine innovative Lösung vor: EvoLLama.Dies ist ein Framework, das Proteinstruktur-Encoder, Sequenz-Encoder und ein großes Sprachmodell für die multimodale Fusion integriert. In der Zero-Shot-Einstellung zeigt EvoLLama starke Generalisierungsfähigkeiten, verbessert die Leistung anderer feinabgestimmter Basismodelle um 1%-8% und übertrifft die durchschnittliche Leistung der aktuellen hochmodernen überwachten Feinabstimmungsmodelle um 6%. Die entsprechenden Forschungsergebnisse wurden als Preprint auf arXiv unter dem Titel „EvoLlama: Enhancing LLMs‘ Understanding of Proteins via Multimodal Structure and Sequence Representations“ veröffentlicht.
Natürlich ist multimodales Lernen nur eine der verfügbaren Forschungsoptionen. Durch den Einsatz weiterer Methoden des maschinellen Lernens zur Untersuchung der Proteinoberfläche können Biologen künftig ein tieferes Verständnis davon gewinnen, wie Proteine mit anderen biologischen Molekülen interagieren, und so die Entwicklung neuer Medikamente unterstützen.
Quellen:
1.https://arxiv.org/abs/2409.03773
2.https://www.frcbs.tsinghua.edu.cn/index.php?c=show&id=873
3.https://www.sohu.com/a/846589543_121124715