Command Palette
Search for a command to run...
Ausgewählt Für ICLR 2025! Shen Chunhua Und Andere Von Der Zhejiang-Universität Schlugen Die Boltzmann-Ausrichtungstechnologie Vor, Und Die Vorhersage Der Freien Energie Der Proteinbindung Erreichte SOTA

Protein-Protein-Interaktionen (PPIs) sind die Grundlage für die Ausführung verschiedener biologischer Funktionen aller Organismen, die hauptsächlich durch die Interaktion und Beeinflussung verschiedener Proteinmoleküle zwischen ihnen erreicht werden. Die genaue Identifizierung und das Verständnis von Protein-Protein-Interaktionen ist äußerst wichtig für die Entschlüsselung von Proteinfunktionen, die Aufklärung von Lebensvorgängen, die Erforschung von Krankheitsmechanismen, die Entwicklung zielgerichteter Medikamente und die Innovation biologischer Anwendungen.
Mit der Entwicklung von Computern und künstlicher Intelligenz hat die Forschung zu PPIs in der wissenschaftlichen Forschungsgemeinschaft in den letzten Jahren mit Unterstützung des Deep Learning große Fortschritte gemacht. Insbesondere AlphaFold 3, das 2024 von DeepMind veröffentlicht wurde,Die Erfolgsrate bei der Vorhersage der Struktur allgemeiner Proteinkomplexe wurde auf fast 80% erhöht.Dadurch wird auch das Problem der hochpräzisen computergestützten Modellierung von Proteininteraktionen gelöst, das die wissenschaftliche Forschungsgemeinschaft seit Jahrzehnten plagt.
Die Interaktion zwischen Proteinen ist jedoch ein dynamischer Prozess, der Bindung und Dissoziation umfasst. Es ist schwierig, die Wechselwirkung zwischen biologischen Molekülen vollständig zu erfassen, wenn man nur statische Strukturen untersucht.Parameter wie die freie Bindungsenergie (∆G, die Differenz der Gibbs-freien Energie zwischen gebundenen und ungebundenen Zuständen) können die Dynamik von Protein-Protein-Interaktionen quantitativ charakterisieren.Die genaue Vorhersage der Änderung der freien Bindungsenergie (∆∆G, auch als Mutationseffekt bekannt) ist jedoch für die wissenschaftliche Gemeinschaft zu einer der Voraussetzungen geworden, um Protein-Protein-Interaktionen zu verstehen oder zu regulieren.
Auf dieser Grundlage hat das Team von Professor Shen Chunhua von der School of Computer Science and Technology der Zhejiang University zusammen mit Teams der University of Adelaide in Australien und der Northeastern University in den Vereinigten StaatenWir schlagen gemeinsam eine Technik namens Boltzmann-Ausrichtung vor, um Wissen aus einem vortrainierten inversen Faltungsmodell auf die Vorhersagen von ∆∆G zu übertragen.Die Studie analysierte zunächst die thermodynamische Definition von ∆∆G und verknüpfte die Energie- und Proteinkonformationsverteilung durch die Einführung der Boltzmann-Verteilung, wodurch das Potenzial vorab trainierter probabilistischer Modelle hervorgehoben wurde. Anschließend nutzte das Team den Satz von Bayes, um die direkte Schätzung zu umgehen und schätzte ∆∆G anhand der Log-Likelihood, die das Protein-Reverse-Folding-Modell lieferte. Diese Ableitung liefert eine rationale Erklärung für die hohe Korrelation zwischen Bindungsenergie und Log-Likelihood des inversen Faltungsmodells, die in anderen früheren Experimenten beobachtet wurde.
Im Vergleich zur vorherigen auf inverser Faltung basierenden Methode zeigen die experimentellen Ergebnisse dieser Methode im SKEMPI v2-Datensatz ein überlegenes Niveau.Sein Spearman-Koeffizient erreichte im überwachten und unbeaufsichtigten Zustand 0,5134 bzw. 0,3201.Deutlich höher als die vorherigen SOTA-Methoden von 0,4324 und 0,2632.
Diese Errungenschaft mit dem Titel „Boltzmann-Aligned Inverse Folding Model as a Predictor of Mutational Effects on Protein-Protein Interactions“ wurde in die ICLR 2025 aufgenommen, die wichtigste internationale akademische Konferenz auf dem Gebiet der künstlichen Intelligenz. Es ist erwähnenswert, dass beim diesjährigen ICLR insgesamt 11.565 Einsendungen eingingen und nur 32.08% der Manuskripte angenommen wurden.

Papieradresse:
https://arxiv.org/abs/2410.09543
Empfehlen Sie eine Veranstaltung zum wissenschaftlichen Austausch. Die letzte Einladung zur Live-Übertragung von Meet AI4S erfolgt am 7. März um 12:00 Uhr.Huang Hong, außerordentlicher Professor an der Huazhong University of Science and Technology, Zhou Dongzhan, Nachwuchsforscher am AI for Science Center des Shanghai Artificial Intelligence Laboratory, und Zhou Bingxin, Assistenzforscher am Institute of Natural Sciences der Shanghai Jiao Tong University,Stellen Sie persönliche Erfolge vor und teilen Sie Erfahrungen aus der wissenschaftlichen Forschung.
Deep Learning beschleunigt Paradigmenwechsel bei der Berechnung von Mutationseffekten
Die wissenschaftliche Gemeinschaft untersucht seit langer Zeit die Vorhersage von ∆∆G.Traditionelle Methoden können in zwei Kategorien unterteilt werden: biophysikalische Methoden und statistische Methoden.Unter ihnen simulieren biophysikalische Methoden hauptsächlich durch Energieberechnungen, wie Proteine auf atomarer Ebene interagieren. Statistische Methoden basieren auf Feature Engineering und verwenden hauptsächlich Deskriptoren, um die geometrischen, physikalischen und evolutionären Eigenschaften von Proteinen zu erfassen.
Es besteht kein Zweifel daran, dass die herkömmliche Methode, egal welche verwendet wird, in hohem Maße auf menschliches Fachwissen angewiesen ist. Dies ist nicht nur zeit- und arbeitsintensiv, sondern auch nicht in der Lage, die komplexen Wechselwirkungen zwischen Proteinen genau zu erfassen. Darüber hinaus haben beide Methoden ihre eigenen Nachteile. Beispielsweise stehen biophysikalische Methoden häufig vor der Herausforderung, Geschwindigkeit und Genauigkeit in Einklang zu bringen. Die auf Deep Learning basierenden Methoden zeigen nicht nur großes „Talent“ in der Proteinmodellierung, sondern beschleunigen auch die Transformation des ∆∆G-Vorhersageparadigmas.
Immer mehr Fälle belegen dies. Beispielsweise hat ein Team der Chinesischen Akademie der Wissenschaften eine auf Repräsentationslernen basierende Methode namens SidechainDiff vorgeschlagen.Diese Methode verwendet das Riemann-Diffusionsmodell, um den Entstehungsprozess von Seitenkettenkonformationen zu erlernen und kann auch eine strukturelle Hintergrunddarstellung von Mutationen an der Protein-Protein-Grenzfläche liefern.Mithilfe der erlernten Darstellungen erreicht die Methode eine hochmoderne Leistung bei der Vorhersage der Auswirkungen von Mutationen auf die Protein-Protein-Bindung.
Dieses Ergebnis trägt den Titel „Vorhersage von Mutationseffekten auf die Protein-Protein-Bindung über ein probabilistisches Modell zur Seitenkettendiffusion“ und ist in NeurIPS 2023 enthalten.
* Papieradresse:
Obwohl mit Deep Learning-basierten Methoden beachtliche Ergebnisse erzielt wurden, sind sie nicht perfekt. Zufälligerweise mit dem obigen Beispiel,In diesem Artikel wird auch erwähnt, dass „es an experimentellen Daten zur Erklärung der Bindungsenergie mangelt“.Dies wird im Allgemeinen als große Herausforderung bei Deep-Learning-Methoden angesehen, was direkt dazu geführt hat, dass mehr Teams dazu neigen, vorab eine große Anzahl nicht gekennzeichneter Datensätze zu trainieren, bevor sie ihre Fähigkeit zur Vorhersage von Mutationen verbessern. Dies umfasst eine Vielzahl von Aufgaben für den Agenten vor dem Training, wie etwa die inverse Proteinfaltung, Maskenmodellierung und Seitenkettenmodellierung im obigen Beispiel.
Diese „alternativen“ Methoden haben zum Glück ihre Ziele erreicht, leider haben sie aber auch ausnahmslos ihre Schwächen gezeigt. Die meisten auf Vortraining basierenden Methoden verwenden nur überwachtes Feintuning (SFT).Die Bedeutung der Datenausrichtung wird jedoch ignoriert, was dazu führen kann, dass das Modell bei der überwachten Feinabstimmung das zuvor während des unbeaufsichtigten Vortrainings erworbene allgemeine Wissen vergisst, was zu einer Überanpassung führt.Rückblickend unterstreichen diese „alternativen“ Methoden zweifellos die Dringlichkeit der Übertragung des erworbenen Wissens für eine genaue Mutationsvorhersage.
Innovative Entwicklung der Boltzmann-Ausrichtung zur Überwindung von SOTA-Modellen
Konkret basierte das Forschungsteam zunächst auf der Boltzmann-Verteilung und den Prinzipien des thermodynamischen Zyklus,Die Änderung der freien Bindungsenergie bei einer Mutation eines Proteins hängt mit der Möglichkeit zusammen, dass die Aminosäuresequenz des Proteins verändert wird.Es wurde die Boltzmann-Ausrichtung vorgeschlagen (wie auf der rechten Seite der Abbildung unten gezeigt). Anschließend schlug das Forschungsteam eine Methode namens BA-Cycle vor, die das inverse Faltungsmodell in die Boltzmann-Ausrichtung integrierte und das inverse Faltungsmodell zur Bewertung von Mutationen durch Vorhersage der Wahrscheinlichkeit von Proteinsequenzen verwendete (wie auf der linken Seite der Abbildung unten gezeigt).
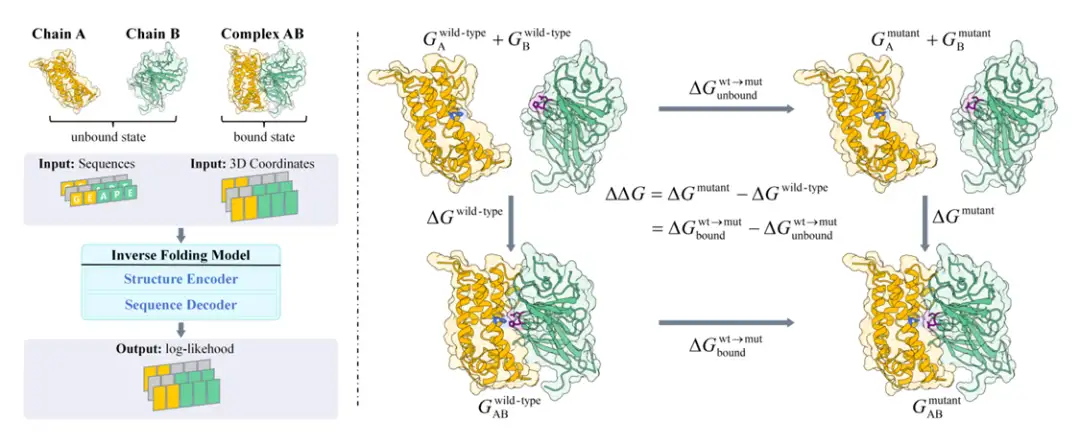
Es ist erwähnenswert, dass zur Herstellung der Verbindung zwischen der freien Energie der Proteinbindung und der bedingten Wahrscheinlichkeit der Proteinsequenz und zur Lösung der beiden Hauptschwierigkeiten bei der direkten Schätzung der Wahrscheinlichkeit p(X|S) der Proteinstruktur unter einer gegebenen Sequenz Folgendes gilt:Die Einschränkungen bestehender Modelle zur Vorhersage der Proteinstruktur und die Unzulänglichkeiten probabilistischer Modelle,Das Forschungsteam ersetzte den Satz von Bayes in die Berechnungsformel für die Bindungsfreiheit, d. h. p(X|S) = p(S|X) ・ p(X)/p(S), und verknüpfte erfolgreich die freie Bindungsenergie mit der bedingten Wahrscheinlichkeit p(X|S) der Proteinsequenz, wodurch die Schwierigkeit der direkten Schätzung von p(X|S) vermieden wurde. Dies legte den Grundstein für die weitere Analyse der Beziehung zwischen Änderungen der freien Bindungsenergie und der bedingten Wahrscheinlichkeit von Proteinsequenzen.
Da man davon ausgeht, dass die Proteinstruktur vor und nach der Mutation unverändert bleibt,Das Forschungsteam verwendete das umgekehrte Faltungsmodell, um die Sequenzwahrscheinlichkeiten gebundener und ungebundener Zustände zu bewerten.Die Grundstruktur des gebundenen Zustands ist normalerweise bekannt und das Modell kann seine Wahrscheinlichkeit direkt berechnen. Die Grundgerüststruktur des ungebundenen Zustands ist nicht explizit angegeben und die Wahrscheinlichkeit kann durch separate Auswertung der beiden Ketten im Komplex geschätzt werden.
Auf dieser GrundlageDas Forschungsteam schlug eine Methode namens BA-Cycle zur unbeaufsichtigten Schätzung von ∆∆G vor.Die unbeaufsichtigte Auswertung von ∆∆G wurde mithilfe des vortrainierten Reverse-Folding-Modells ProteinMPNN durchgeführt. Dies steht in starkem Kontrast zu früheren Studien, die die Wahrscheinlichkeit ungebundener Zustände im thermodynamischen Zyklus nicht explizit berücksichtigten.
endlich,Das Forschungsteam schlug außerdem eine Methode namens BA-DDG vor.Der BA-Zyklus wurde durch Boltzmann-Ausrichtung unter Verwendung der Daten zur Änderung der Bindungs-Freienergiebezeichnung feinabgestimmt. BA-DDG verwendet denselben Vorwärtsprozess wie BA-Cycle. Das Ziel von BA-DDG besteht darin, die Lücke zwischen der tatsächlichen Änderung der Bindungs-Freienergie und der vorhergesagten Änderung der Bindungs-Freienergie zu minimieren und gleichzeitig die Verteilung des ursprünglichen vortrainierten Modells beizubehalten.
Das Forschungsteam führte eine Reihe experimenteller Überprüfungen des SKEMPI v2-Datensatzes durch.Darunter ist der SKEMPI v2-Datensatz ein annotierter Mutationsdatensatz, der 348 Proteinkomplexe enthält, darunter 7.085 Aminosäuremutationen sowie Änderungen der thermodynamischen Parameter und kinetischen Geschwindigkeitskonstanten.
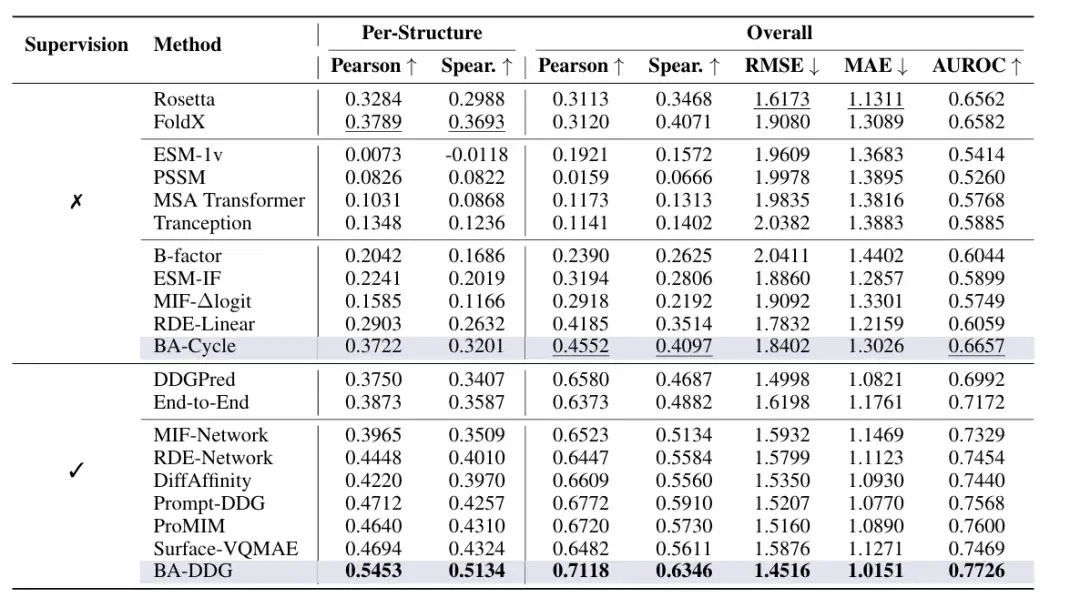
Insgesamt gibt es 7 Bewertungsindikatoren, darunter 5 Gesamtindikatoren, nämlich den Pearson-Korrelationskoeffizienten, den Spearman-Rangkorrelationskoeffizienten, den minimalen mittleren quadratischen Fehler (RMSE), den minimalen mittleren absoluten Fehler (MAE) und AUROC. Darüber hinaus gruppierte das Forschungsteam die Mutationen nach ihren strukturellen Merkmalen und berechnete für jede Gruppe den Pearson-Korrelationskoeffizienten und den Spearman-Korrelationskoeffizienten als zwei zusätzliche Indikatoren.
Das Forschungsteam verglich zunächst BA-Cyale und BA-DDG mit den unbeaufsichtigten und überwachten Methoden von SOTA.Es gibt drei Arten von unüberwachten Methoden, darunter traditionelle empirische Energiefunktionen wie Rosetta Cartesian ∆∆G und FoldX; sequenz-/evolutionsbasierte Methoden wie ESM-1v, Position-Specific Scoring Matrix (PSSM), MSA Transformer und Tranception; und vortrainierte Methoden basierend auf Strukturinformationen, die nicht auf ∆∆G-Labels trainiert sind, wie ESM-1F, MIF-∆logits, RDE-Linear und B-Faktor.
Überwachte Methoden werden in zwei Kategorien unterteilt, darunter End-to-End-Lernmodelle wie DDGPred und End-to-End; und Vortrainingsmethoden basierend auf Strukturinformationen, fein abgestimmt auf ∆∆G, einschließlich MIF-Network, RDE-Network, DiffAffinity, Prompt-DDG, ProMIM und Surface-VQMAE.
Die Ergebnisse zeigen, dassBA-DDG übertrifft alle Baselines in allen Bewertungsmetriken.Unter ihnen erreichten der Pearson-Korrelationskoeffizient und der Spearman-Korrelationskoeffizient unter der überwachten Methode 0,5453 bzw. 0,5134. Die deutliche Verbesserung der Korrelation der einzelnen Strukturen unterstreicht die höhere Zuverlässigkeit in praktischen Anwendungen.BA-Cycle erreicht eine vergleichbare Leistung wie die empirische Energiefunktion und übertrifft alle unüberwachten Lern-Baselines.Wie in der folgenden Abbildung dargestellt:
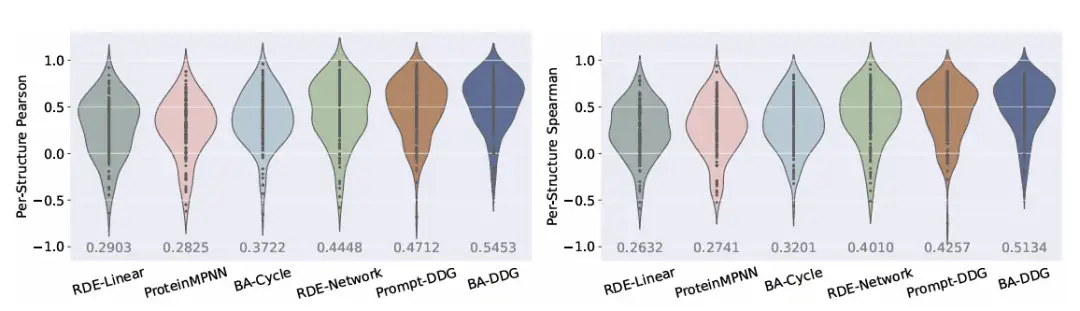
Darüber hinaus ist in der entsprechenden visuellen Analyse klar ersichtlich, dassBA-DDG übertrifft andere Methoden sowohl bei der qualitativen Visualisierung als auch bei den quantitativen Messwerten.Wie in der folgenden Abbildung dargestellt:
Darüber hinaus führten die Forscher Experimente zur Vorhersage der Bindungsenergie, zum Protein-Protein-Docking und zur Antikörperoptimierung durch, und die Ergebnisse zeigten deren breite Anwendbarkeit. Diese positiven Auswirkungen werden bei der Entwicklung von Arzneimitteln und beim virtuellen Screening eine äußerst wichtige Rolle spielen und eine theoretische Grundlage für ihre tatsächliche Anwendung in der Zukunft legen.
Umfassende Entwicklung von maschinellem Lernen und maschinellem Sehen zur Realisierung der KI-Universalisierung
In dieser Studie nutzten die Forscher fachübergreifende Theorien, um neue Perspektiven für die Protein-Sequenzanalyse zu bieten, und schufen gleichzeitig durch innovative Modellintegration und Modelloptimierung einen systematischen Forschungsrahmen. Diese schrittweise Forschungsmethode hilft nicht nur dabei, die Beziehung zwischen Proteinsequenz und Änderungen der freien Energie vollständig und gründlich zu verstehen, sondern liefert auch neue Ideen für nachfolgende Forschungen.
Es ist erwähnenswert, dassProfessor Shen Chunhua, einer der Hauptteilnehmer dieser Forschung, engagiert sich seit langem in der Erforschung des maschinellen Lernens und der Computervision.Er hat bisher mehr als 150 Artikel veröffentlicht, darunter einige auf international renommierten akademischen Plattformen wie TPAMI und IJCV. Nur zwei Monate nach Beginn des Jahres 2025 hat das von Professor Shen Chunhua geleitete Team wichtige Ergebnisse erzielt und drei Artikel auf der Preprint-Plattform arXiv veröffentlicht.
Im ersten Artikel entwickelte die Forschungsgruppe von Professor Shen Chunhua ein DNA-basiertes Modell namens ConvNova, das auf dem CNN-Netzwerk basiert. Das Modell ist einfach im Design, hat aber eine bemerkenswerte Leistung.Bei der zugehörigen Histon-Aufgabe übertraf die Durchschnittspunktzahl die zweitplatzierte Methode 5.8%, wodurch schnellere Berechnungen mit weniger Parametern erreicht wurden.Gleichzeitig bestätigt diese Methode auch, dass die auf der CNN-Netzwerkarchitektur basierende Methode im Vergleich zu den auf dem Transformer-Netzwerk und dem SSM-Netzwerk basierenden Methoden ein starkes Wettbewerbspotenzial hat. Die zugehörige Forschung wurde unter dem Titel „Revisiting Convolution Architecture in the Realm of DNA Foundation Models“ veröffentlicht.
* Papieradresse:
https://arxiv.org/abs/2502.18538
Im zweiten Artikel entwickelten die Forschungsgruppe von Professor Shen Chunhua und das Shanghai AI Laboratory gemeinsam ein allgemeines Sehmodell DICEPTION.Das vortrainierte Diffusionsmodell wird zur Lösung von Multitasking-Problemen der visuellen Wahrnehmung verwendet, erfordert weniger Trainingsdaten und weist eine starke Anpassungsfähigkeit an Aufgaben auf.Mit nur 0,061 TP3T an SAM-Daten erreicht das Modell bei Aufgaben wie der Segmentierung ein mit SOTA-Modellen vergleichbares Niveau und reduziert die Trainingskosten erheblich, indem es die Aufgabenausgaben durch Farbcodierung vereinheitlicht. Die zugehörige Forschung wurde unter dem Titel „DICEPTION: A Generalist Diffusion Model for Visual Perceptual Tasks“ veröffentlicht.
* Papieradresse:
https://arxiv.org/pdf/2502.17157
Im dritten Artikel schlug das Team von Professor Shen Chunhua in Zusammenarbeit mit Alibaba einen Benchmark namens PhyCoBench vor, der verwendet wird, um die Fähigkeit von Videogenerierungsmodellen zu bewerten, Videos zu generieren, die den Gesetzen der Physik entsprechen. Die Studie stellt außerdem das automatische Auswertungsmodell PhyCoPredictor vor, ein Diffusionsmodell, das optischen Fluss und Videobilder kaskadenartig generiert. Durch den Vergleich der Konsistenzbewertung der automatischen und manuellen Sortierung,Experimentelle Ergebnisse zeigen, dass PhyCoPredictor über die Fähigkeiten verfügt, die der menschlichen Bewertung am nächsten kommen.Die zugehörige Forschung wurde unter dem Titel „A Physical Coherence Benchmark for Evaluating Video Generation Models via Optical Flow-guided Frame Prediction“ veröffentlicht.
* Papieradresse:
https://arxiv.org/pdf/2502.05503
Das Team von Professor Shen Chunhua hat nicht nur fruchtbare Ergebnisse erzielt, auch sein persönlicher Einfluss ist herausragend. Die von Professor Shen Chunhua veröffentlichten einschlägigen Arbeiten waren schon immer eine wichtige Quelle für Zitate in der wissenschaftlichen Forschungsgemeinschaft. Er wurde außerdem in die Liste „2023 Highly Cited Chinese Researchers“ von Elsevier, einem globalen Informationsanalyseunternehmen, aufgenommen.
Heute ist Professor Shen Chunhua seit drei Jahren Qiushi-Lehrstuhlprofessor und stellvertretender Direktor des National Key Laboratory of Computer-Aided Design and Image Systems an der Zhejiang-Universität. Er hat nicht nur fruchtbare Forschungsergebnisse erzielt, sondern auch beachtliche Lehrleistungen erbracht und zahlreiche Master- und Doktoranden ausgebildet. Darüber hinaus dient das dort angesiedelte Nationale Schlüssellabor für computergestütztes Design und Grafiksysteme als Schnittstelle zwischen „Industrie, Universität und Forschung“ und hat in den letzten Jahren ebenfalls eine vielschichtige Entwicklung durchlaufen. Es hat mit vielen Unternehmen, darunter Ant, zusammengearbeitet und sich zu einer Innovationsbasis für wissenschaftliche Forschung, einer Talentausbildungsbasis und einer Innovationsinkubationsbasis entwickelt.








