Command Palette
Search for a command to run...
Im Nature Journal Veröffentlicht! Das Team Der Peking-Universität Nutzt KI, Um Die Evolutionsrichtung Von COVID-19-/AIDS-/Influenzaviren Vorherzusagen, Mit Einer Genauigkeitsverbesserung Von 67%
Im Dezember 2019 brach plötzlich die COVID-19-Pandemie aus. Diese durch das SARS-CoV-2-Virus verursachte Krankheit ist hoch ansteckend. Innerhalb nur eines Monats überstieg die Zahl der Fälle in meinem Land 1.000 und verbreitete sich rasch auf der ganzen Welt.
Um die weitere Ausbreitung des Virus zu bekämpfen, hat unser Land Anfang 2021 eine allgemeine kostenlose Impfpolitik eingeführt. Doch selbst mit der Unterstützung von Impfstoffen hat sich die Gesundheitskrise zunehmend verschärft.Dies liegt daran, dass das SARS-CoV-2-Virus weiterhin mutiert.Durch die allmähliche Anpassung an den durch den Impfstoff erzeugten Immundruck und die veränderten Umweltbedingungen ist der ursprünglich in Wuhan entdeckte „Virusstamm“ längst verschwunden und durch verschiedene mutierte Stämme ersetzt worden, die weiterhin neue Infektionswellen auslösen und deren Auswirkungen noch nach 2023 anhalten werden.
Zufälligerweise ist die Influenzavirus-Positivitätsrate erst kürzlich weiter gestiegen und viele Menschen haben sich mit Influenza A (A19) infiziert, ohne es zu wissen. Ähnlich wie das SARS-CoV-2-Virus ist auch A19 hoch ansteckend, verbreitet sich schnell und mutiert rasch. In derselben Saison können mehrere Subtypen des Virus auftreten, wodurch sich auch das Risiko einer erneuten Infektion der Bevölkerung innerhalb kurzer Zeit erhöht.
Dies zeigt, dass die Vorhersage der Richtung der Virusentwicklung für die Prävention und Kontrolle sowie die Entwicklung von Impfstoffen und Medikamenten von entscheidender Bedeutung ist.Da Mutationen jedoch die Grundlage der Virusevolution bilden, sind sie höchst zufällig, sodass in der Regel nur eine sehr kleine Zahl von Mutationen die Anpassungsfähigkeit des Virus „gerade so“ erhöhen kann. Dieses Ungleichgewicht zwischen positiven Proben (vorteilhaften Mutationen) und negativen Proben (schädlichen Mutationen) macht es äußerst schwierig, ein Deep-Learning-Modell zu trainieren, das seltene vorteilhafte Mutationen des Virus vorhersagen kann. Gleichzeitig mutieren Viren oft nur an wenigen Stellen, was es für neuronale Netzwerke schwierig macht, die schwachen Veränderungen der intramolekularen Wechselwirkungen, die durch Mutationen verursacht werden, direkt zu erfassen, und auch Probleme bei der Modellierung mit sich bringt.
In diesem Zusammenhang leiteten Professor Tian Yonghong und Associate Professor Chen Jie von der School of Information Engineering der Peking-Universität zusammen mit dem Forscher Zhou Peng vom Guangzhou National Laboratory den Doktoranden Nie Zhiwei und den Masterstudenten Liu Xudong an, das Problem der Vorhersage der viralen Evolution erneut zu untersuchen und schlugen ein evolutionsbasiertes Rahmenwerk zur Vorhersage der treibenden Kraft von Virusmutationen, E2VD, vor.Mithilfe dieses Rahmens lässt sich die evolutionäre Richtung des SARS-CoV-2-Virus, des Influenzavirus, des Zika-Virus und des HIV (AIDS-Virus) vorhersagen, wodurch die Geschwindigkeit der menschlichen Reaktion auf neu auftretende Virusinfektionen deutlich verbessert und eine wichtige Unterstützung für die schnelle Optimierung von Impfstoffen und Medikamenten geleistet werden kann.
Die Forschungsergebnisse wurden am 17. Januar 2025 in Nature Machine Intelligence unter dem Titel „Ein einheitliches, evolutionsbasiertes Deep-Learning-Framework zur Vorhersage von Virusvariationstreibern“ veröffentlicht.

Papieradresse:
https://www.nature.com/articles/s42256-024-00966-9
Papieradresse: Folgen Sie dem offiziellen Konto und antworten Sie mit „Viral Evolution“, um das vollständige PDF zu erhalten
Das Open-Source-Projekt „awesome-ai4s“ vereint mehr als 100 AI4S-Papierinterpretationen und stellt umfangreiche Datensätze und Tools bereit:
https://github.com/hyperai/awesome-ai4s
Datensatz: UniRef90-Vortrainingsdatensatz und Datensatz zum tiefen Mutationsscannen von Viren
Viren erzeugen im Laufe der Evolution kontinuierlich neue Mutationen und akkumulieren diese selektiv. Daher muss das Proteinsprachenmodell für Evolutionsszenarien über starke Zero-Sample-Generalisierungsfähigkeiten verfügen, d. h., es muss in der Lage sein, mit ungesehenen Mutationen umzugehen. Um dies zu erreichen,Das Forschungsteam wählte UniRef90 als Datensatz für das Vortraining des Proteinsprachenmodells. UniRef90 enthält umfangreiche evolutionäre Informationen auf Sequenzebene, ohne die Leistung in den frühen Phasen des Modelltrainings negativ zu beeinflussen. Diese umfangreichen evolutionären Informationen ermöglichen es, das Modell während des Vortrainings einer ausreichenden Anzahl von Proteinfamiliensequenzproben auszusetzen und so seine Fähigkeit zur Nullprobengeneralisierung zu verbessern.
Um das Modell beim Erlernen der evolutionären Fitnesslandschaft zu unterstützen, die durch virale Mutationen verursacht wird,Das Forschungsteam verwendete Open-Source-Datensätze zum Deep Mutation Scanning verschiedener Viren.
Modellarchitektur: Evolutionsinspiriertes universelles Architekturdesign
Basierend auf den Konzepten der „schwachen Mutationsverstärkung“ und des „Mining seltener vorteilhafter Mutationen“ schlug das Forschungsteam das evolutionsbasierte Rahmenwerk E2VD zur Vorhersage der treibenden Kraft viraler Mutationen vor. Wie in Abbildung a unten gezeigt,Es umfasst hauptsächlich 3 Module:Dabei handelt es sich um die Protein-Sequenzkodierung, die lokale-globale Abhängigkeitskopplung und das fokale Lernen mehrerer Aufgaben.
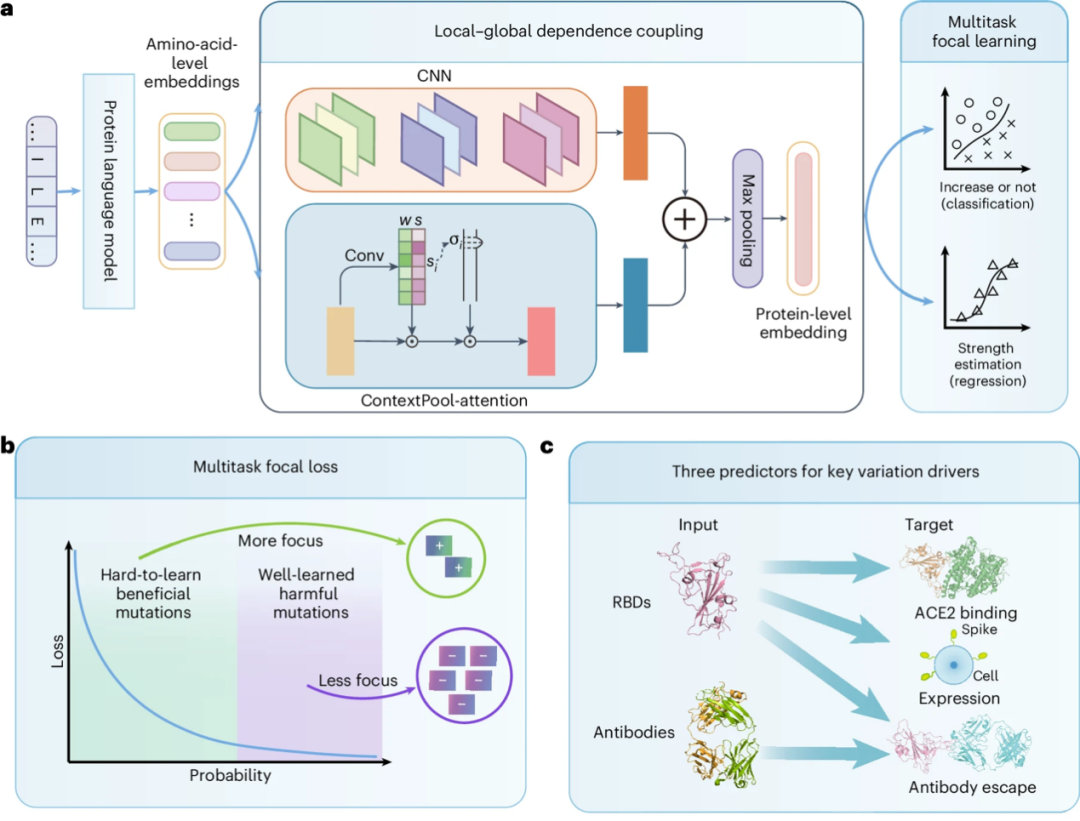
* Erste,Im Modul zur Protein-Sequenz-Codierung trainierte das Forschungsteam eigenständig ein maßgeschneidertes Proteinsprachenmodell für die virale Evolution, das die Eigenschaften viraler Proteinsequenzen präzise extrahieren kann.
* Zweitens,Im Modul zur Fusion lokaler und globaler Interaktionsabhängigkeiten verwendeten die Forscher Convolutional Neural Networks (CNNs), um die Interaktionsabhängigkeiten zwischen Mutationen und benachbarten Aminosäuren zu erfassen, und entwarfen einen lernbaren dynamischen Aufmerksamkeitsmechanismus, um ein weitreichendes Interaktionsabhängigkeitsnetzwerk auf der Motivebene zu konstruieren, auf der sich die Mutation befindet. Dieses Design löst effektiv das Problem, dass schwache Effekte, die durch weniger Mutationen insgesamt in der Variante verursacht werden, schwer zu erfassen sind.
* Dann,Im Multi-Task-Fokus-Lernmodul werden die Vorteile des Multi-Task-Lernens und schwieriger Sample-Mining-Strategien kombiniert, um die Vorhersageleistung des Modells für die Fitness von Virusmutationen durch die gemeinsame Nutzung von Parametern beim Multi-Task-Training zu verbessern.
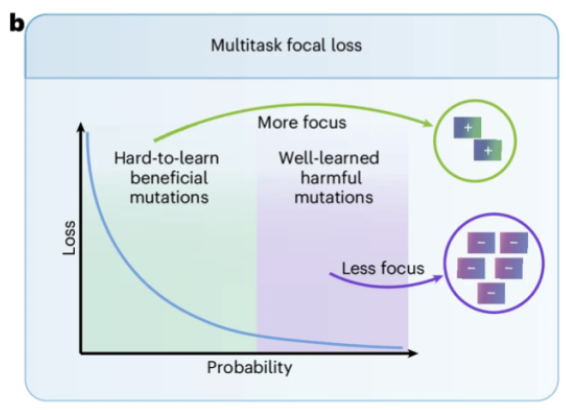
Noch wichtiger ist, dass das Team, wie in Abbildung b oben gezeigt, eine neuartige Multitasking-Fokusverlustfunktion entwickelt hat, die das Modell dazu veranlasst, seltenen vorteilhaften Mutationen, die während des Trainings nur schwer effektiv erlernbar sind, mehr Aufmerksamkeit zu schenken, wodurch die Vorhersageleistung für seltene vorteilhafte Mutationen (d. h. schwierige Proben) erheblich verbessert wird.
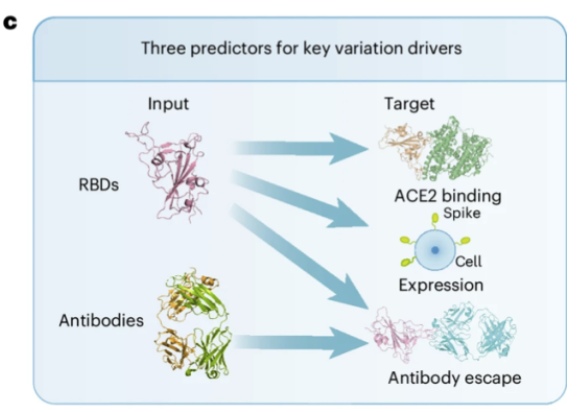
Darüber hinaus kann das E2VD-Vorhersageframework, wie in Abbildung c oben gezeigt, die Eingabe und Ausgabe für verschiedene Aufgaben zur Vorhersage der Virusfitness flexibel anpassen. Um beispielsweise durch Mutationen verursachte Änderungen der Bindungsaffinität vorherzusagen, kann nur die Virussequenz eingegeben werden. Um durch Mutationen verursachte Änderungen der Fähigkeit von Antikörpern, zu entkommen, vorherzusagen, können sowohl die Virussequenz als auch die Antikörpersequenz usw. eingegeben werden. Dadurch können hochpräzise evolutionäre Vorhersagen für alle Virustypen und -stämme auf einer einheitlichen Architektur erreicht werden.
Konkret wurde in der Studie das E2VD-Framework für Vorhersageaufgaben im Zusammenhang mit dem SARS-CoV-2-Virus, Influenza (Grippevirus), Zika (Zika-Virus) und HIV (AIDS-Virus) verwendet:
* Zu den Aufgaben im Zusammenhang mit SARS-CoV-2 gehört die Vorhersage der Bindungsaffinität, Expression und Antikörperflucht, die die Haupttreiber der Virusmutation sind.
* Die Aufgabe für Influenza-, Zika- und HIV-Viren besteht darin, den durch Mutationen verursachten Fitnesseffekt vorherzusagen, um die Generalisierungsfähigkeit des Modells zu analysieren.
Experimentelle Ergebnisse: E2VD verbessert die Genauigkeit der Vorhersage vorteilhafter Mutationen durch 67% und weist eine hervorragende Generalisierungsleistung auf
E2VD kann virale Evolutionsmuster präzise erfassen und die Genauigkeit der Vorhersage vorteilhafter Mutationen durch 67% verbessern
Das Team verglich die Vorhersageleistung der angepassten Proteinsprache für evolutionäre Szenarien mit der des gängigen Proteinsprachenmodells. Die Ergebnisse zeigten, dass das maßgeschneiderte Proteinsprachenmodell des Teams mit mindestens 340 Millionen Modellparametern die beste Vorhersageleistung erzielte und sogar ESM2-15B übertraf, das über die 44-fache Anzahl an Parametern verfügt. Dies beweist die Wirksamkeit des angepassten Vortrainingsdatensatzes und der Trainingsstrategie.
Anschließend verglich das Team E2VD mit gängigen Methoden bei verschiedenen Aufgaben zur Vorhersage wichtiger viraler evolutionärer Antriebskräfte. Die Ergebnisse zeigten, dass E2VD andere Methoden deutlich übertraf, mit Leistungsverbesserungen im Bereich von 7% bis 21%. Um die Fähigkeit von E2VD zu demonstrieren, virale Evolutionsmuster präzise zu erfassen, beispielsweise verschiedene Mutationstypen präzise zu unterscheiden und seltene vorteilhafte Mutationen präzise herauszufiltern, führten die Forscher außerdem mehrere Experimente durch.
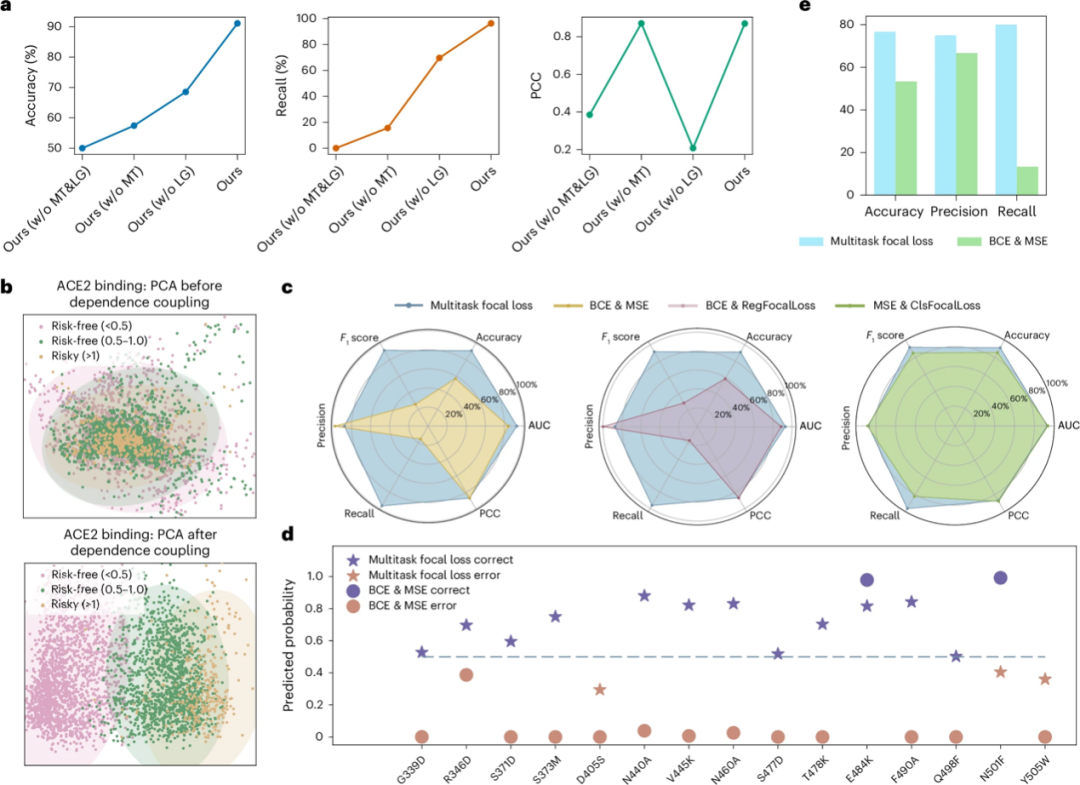
a: ohne MT bedeutet E2VD ohne MT-Modul; ohne LG bedeutet E2VD ohne LG-Modul; ohne MT&LG bedeutet E2VD ohne MT&LG-Modul
b: Drei Mutationstypen mit Risikostufen, die in der Aufgabe zur Vorhersage der Bindungsaffinität beschrieben wurden
d: Die Fähigkeit verschiedener Verluste, seltene vorteilhafte Mutationen zu erfassen
Zunächst werden Modulablationsstudien durchgeführt, um die Beiträge des Moduls „Local-Global Interaction Dependency Fusion“ (LG) und des Moduls „Multi-Task Focal Learning“ (MT) zur Vorhersageleistung zu untersuchen. Wie in Abbildung a oben gezeigt, ergab die Studie, dass das MT-Modul beim Mining seltener vorteilhafter Mutationen in der viralen Fitness effektiv ist (die Rückrufrate stieg von 0 auf 69,63%). Durch die Kombination des LG-Moduls mit dem MT-Modul kann die Modellleistung weiter verbessert werden, mit einer Genauigkeit von 91,11%, einem Rückruf von 96,3% und einem Korrelationskoeffizienten von 0,87.
Die vom Team vorgeschlagene Multi-Task-Focal-Loss-Funktion kann die Vorhersageleistung erheblich verbessern. Um die Fähigkeit des Multi-Task-Focal-Loss zur Erfassung seltener vorteilhafter Mutationen zu bewerten, wählten die Forscher repräsentative vorteilhafte und schädliche Mutationen aus, um einen Testsatz zu bilden.
* Im Hinblick auf die Vorhersage vorteilhafter Mutationen verbessert E2VD, wie in Abbildung d oben gezeigt, die Vorhersagegenauigkeit seltener vorteilhafter Mutationen von 13% auf 80% und erreicht damit eine sprunghafte Verbesserung der Genauigkeit. Dadurch werden seltene vorteilhafte Mutationen, die für die virale Evolution entscheidend sind, präzise und effizient ermittelt.
* Bei schädlichen Mutationen weisen Multi-Task-Focal-Loss und herkömmliche BCE&MSE eine ähnliche Leistung auf. Dies liegt daran, dass BCE&MSE dem Modell nicht dabei helfen können, die seltenen vorteilhaften Mutationen zu erlernen, was dazu führt, dass das Modell dazu neigt, alle Mutationen als schädliche Mutationen vorherzusagen.
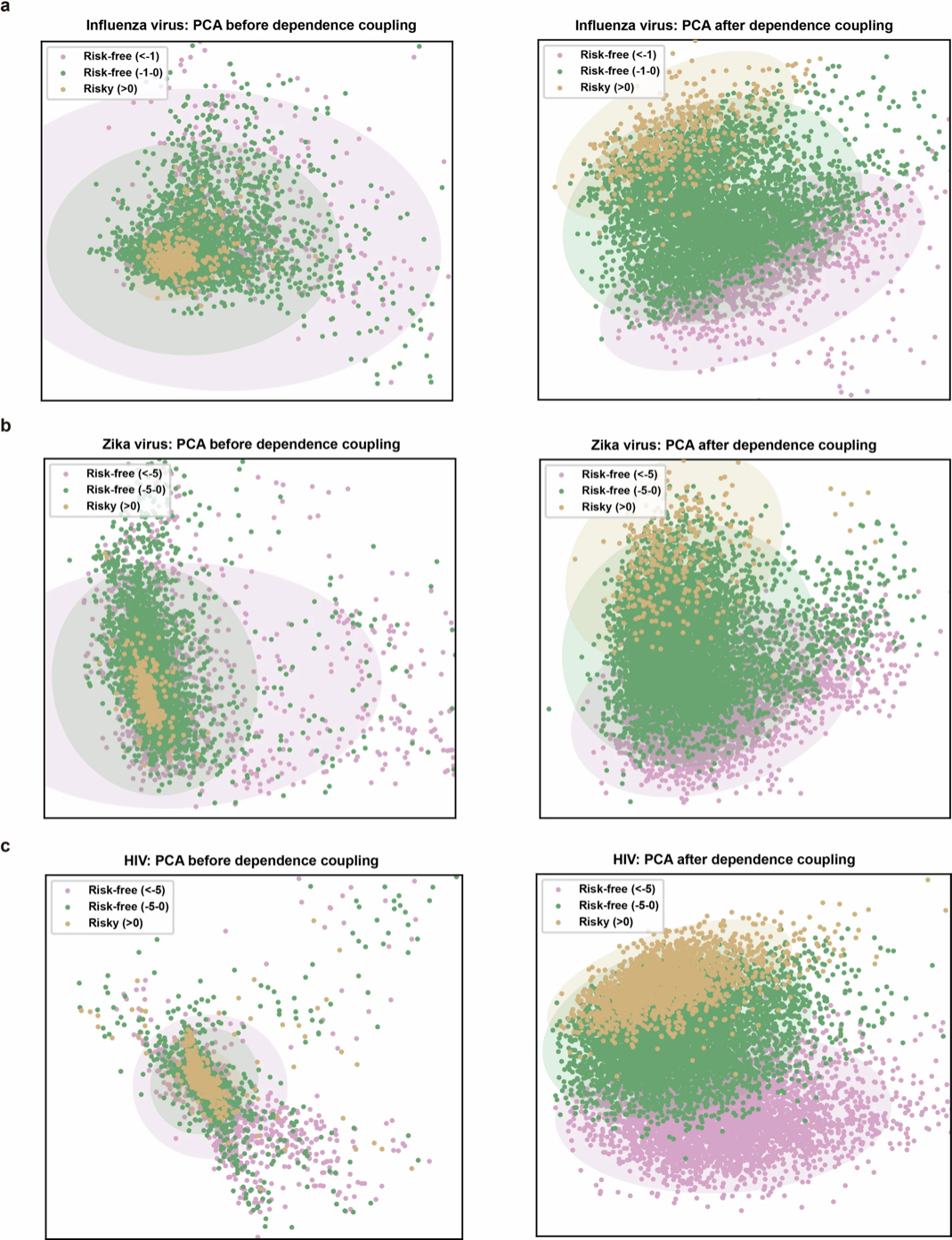
Wie in Abbildung b unten gezeigt, verwendeten die Forscher die Hauptkomponentenanalyse (PCA), um die Dimensionsreduktion von drei Mutationstypen bei Influenza, Zika und HIV zu visualisieren. Die Ergebnisse zeigten, dass nach der Verarbeitung durch das LG-Modul die Merkmale verschiedener Mutationen deutlich unterschieden und klare Grenzen aufwiesen. Dies deutet darauf hin, dass LG die Empfindlichkeit von E2VD gegenüber verschiedenen Mutationstypen erhöhen kann, indem es das intramolekulare Interaktionsnetzwerk erfasst und rekonstruiert und so ein besseres Verständnis der evolutionären Anpassungsfähigkeit des Virus ermöglicht.
E2VD verfügt über eine hervorragende Generalisierungsleistung und kann Vorhersagen über verschiedene Virustypen und -stämme hinweg treffen
Viren entwickeln sich unter Selektionsdruck weiter, was zur Entstehung mehrerer Stämme führen kann. Beispielsweise umfasst das Grippevirus, das in letzter Zeit viel Aufmerksamkeit erregt hat, mehrere Typen und weist saisonale Mutationen auf. Daher ist die Generalisierungsfähigkeit des Modells von entscheidender Bedeutung, um mit komplexen Virusentwicklungstrends fertig zu werden. Die Forscher schlugen „Ordinal Pair Proportion“ (OPP) vor, um die Generalisierungsfähigkeit des Modells bei der Vorhersage verschiedener Stämme desselben Virus und verschiedener Virustypen zu bewerten.
* OPP stellt den Anteil der korrekt vorhergesagten Mutationspaare an allen Mutationspaaren dar. Je höher der OPP-Wert, desto weniger chaotisch ist die vorhergesagte adaptive Landschaft, was darauf hindeutet, dass das Modell die relative Reihenfolge der Treiber viraler Mutationen besser vorhersagen kann.
Wie in Abbildung b unten gezeigt, haben die Forscher für die Aufgabe der Vorhersage der Bindungsaffinität zwischen Stämmen die OPP von 6 verschiedenen Stämmen und allen gemischten Stammdaten (Alle) ausgewertet und festgestellt, dass E2VD in allen Fällen andere Methoden deutlich übertraf. Wie in Abbildung c unten gezeigt, übertrifft E2VD bei der Vorhersage des Expressionsniveaus für die meisten Stämme andere Methoden. Insgesamt übertrifft E2VD modernste Methoden bei Stämmen außerhalb der Verteilung bei weitem und weist eine äußerst generalisierbare Leistung auf.
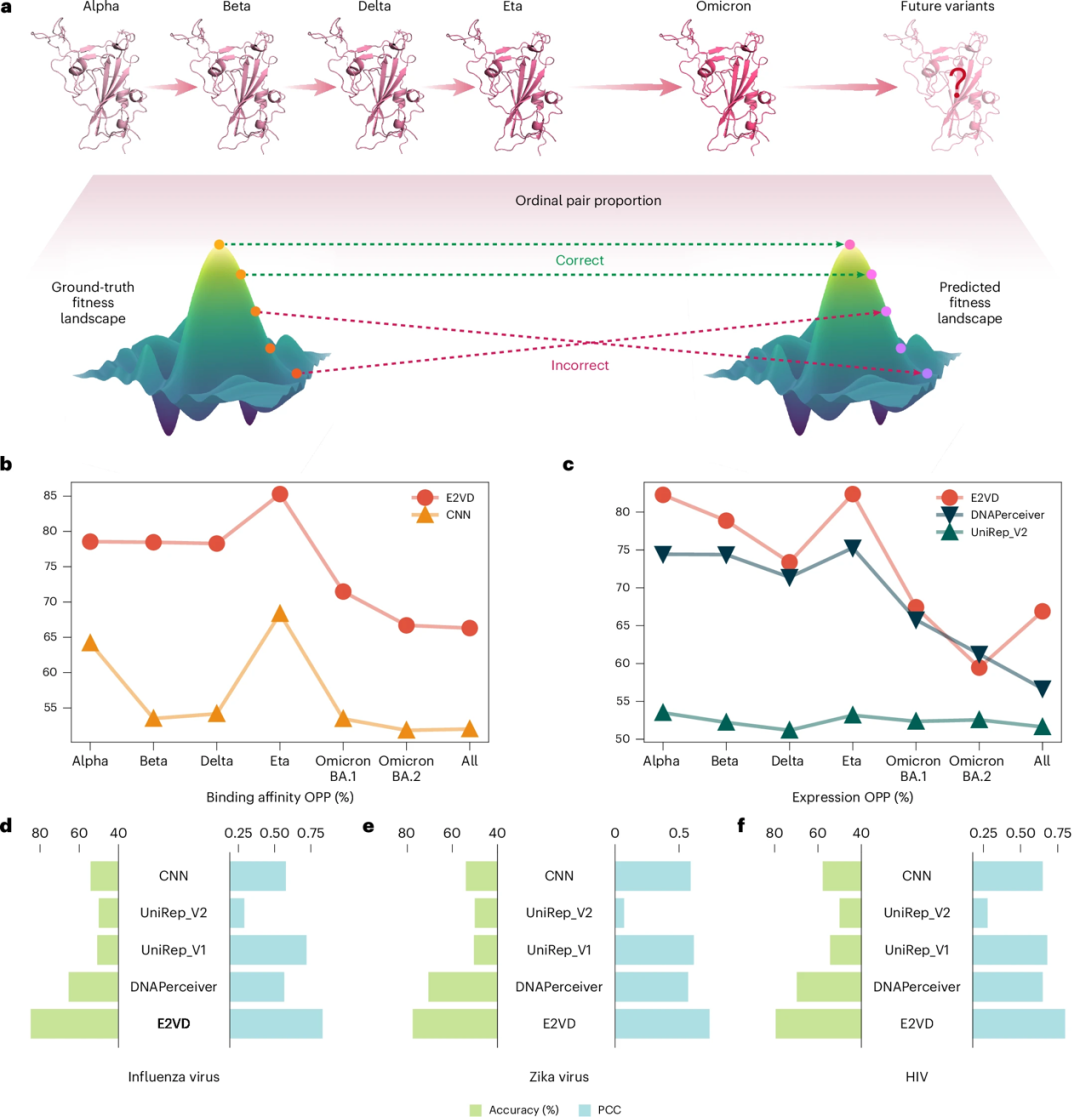
* b, c: E2VD sagt den OPP verschiedener Virusstämme voraus; d, e, f: E2VD sagt die Leistung verschiedener Virentypen voraus
Wie in den Abbildungen d, e und f oben gezeigt, stellten die Forscher bei der Vorhersage verschiedener Virustypen fest, dass E2VD ideale Generalisierungsfähigkeiten für das neue Coronavirus, das Zika-Virus, das Influenzavirus und HIV aufwies, andere Methoden bei weitem übertraf und in Zukunft möglicherweise noch auf infektiösere Viren ausgeweitet werden könnte.
KI hat großes Potenzial zur Vorhersage der Virusentwicklung
Die oben genannte Forschung untersuchte das Problem der Vorhersage der Virusevolution aus der Perspektive der Evolution neu und konstruierte ein universelles Rahmenwerk zur Evolutionsvorhersage (E2VD), das für verschiedene Virustypen und -stämme geeignet ist. Dieses Framework zeigte eine hervorragende Vorhersageleistung und Generalisierungsfähigkeit bei der Vorhersage mehrerer Treiberfaktoren für Virusmutationen, wodurch es möglich wurde, Trends in der Virusentwicklung vorherzusagen.Darüber hinaus kann durch die flexible und maßgeschneiderte Kombination von E2VD auch die Vorhersage von Evolutionstrends auf verschiedenen Skalen realisiert werden.
* Erstens kann E2VD den Verlauf der Virusevolution während Pandemien erklären und uns helfen, die Gründe für die Verbreitung bestimmter Stämme und die ihnen zugrunde liegenden molekularen Mechanismen zu verstehen.
* Zweitens ist E2VD in Kombination mit einer virtuellen Deep-Mutation-Scanning-Simulation in der Lage, mögliche Hochrisikomutationen vorherzusagen und erreicht dabei eine Trefferquote von 80%.
* Schließlich ermöglicht E2VD auch die Vorhersage makroevolutionärer Trajektorien im Pandemiemaßstab, indem es den Evolutionspfad des Virus in der realen Welt reproduziert und so theoretische Unterstützung für die Interpretation des Virusevolutionsmechanismus liefert.
In Zukunft plant das Team, E2VD mit Impfstoff- und Proteinmedikamenten-Designprozessen zu kombinieren, um die Effizienz und Steuerbarkeit des Designs zu verbessern, was für die Virusprävention und -kontrolle sowie das Medikamentendesign von großer Bedeutung sein wird.
Erwähnenswert ist, dass die Autoren der Studie Professor Tian Yonghong und Associate Professor Chen Jie von der School of Information Engineering der Peking-Universität sowie ihre Doktoranden Nie Zhiwei und Masterstudent Liu Xudong sind. Das Team konzentriert sich weiterhin auf die Forschung im Bereich KI für Biowissenschaften. Ihr Projekt „Ahead of the Evolution of the Virus – Predicting Future High-Risk Coronavirus Variants through Artificial Intelligence Simulation“ wurde im November 2022 erfolgreich für den „Gordon Bell New Crown Special Award“ 2022 nominiert (der Gordon Bell Preis ist die weltweit höchste akademische Auszeichnung im Bereich der Hochleistungsrechneranwendungen).
Das Team verfügt über umfassende Erfahrung auf dem Gebiet der Vorhersage der Virusentwicklung. Im Juli 2023 veröffentlichte das Team im International Journal of High Performance Computing Applications „Running ahead of evolution—AI-based simulation for predicting future high-risk SARS-CoV-2 variants“. Konkret trainierten die Forscher vorab ein großes Proteinsprachenmodell und konstruierten eine Hochdurchsatz-Screeningmethode auf Grundlage der Bindungsaffinität und der Vorhersage des Antikörper-Escapes. Dies ist die erste Studie zur Simulation von SARS-CoV-2-RBD-Mutationen. Das Modell konnte erfolgreich Mutationen in der RBD-Region von fünf besorgniserregenden Varianten identifizieren und innerhalb weniger Sekunden Millionen potenzieller Varianten aussortieren. Damit bietet es ein technisches Mittel zur Prävention und Kontrolle von Epidemien in Form eines „KI+HPC“-Paradigmas (künstliche Intelligenz + Hochleistungsrechnen).
Link zum Artikel:
https://journals.sagepub.com/doi/abs/10.1177/10943420231188077
Darüber hinaus hat das Team eine Reihe grundlegender Modelle für die Biowissenschaften entwickelt. Am Beispiel der für die Enzymtechnik entscheidenden Aufgabe der Vorhersage von „Enzym-Substrat“-Interaktionen veröffentlichte das Team im Dezember 2024 einen Vorabdruckartikel, in dem es ein progressives bedingtes Deep-Learning-Framework (MESI) für die Mehrzweckvorhersage von Enzym-Substrat-Interaktionen vorschlägt.
Link zum Artikel:
https://www.researchsquare.com/article/rs-5516445/v1
Insbesondere werden durch die Entkopplung der Modellierung von Enzym-Substrat-Interaktionen in einen zweistufigen Lernprozess zwei bedingte Netzwerke entwickelt, um jeweils die Spezifität der Enzymreaktionen und wichtige Informationen zur katalytischen Interaktion einzuführen und so den allmählichen Übergang des latenten Merkmalsraums vom allgemeinen Bereich der Proteine und kleinen Moleküle zum katalytisch bewussten Bereich zu erleichtern. Das Modell übertrifft bei verschiedenen nachgelagerten Aufgaben stets die modernsten Methoden. Darüber hinaus erfasst das vorgeschlagene bedingte Netzwerk implizit die wesentlichen Arten der Enzymkatalyse mit vernachlässigbarem zusätzlichen Rechenaufwand. Unterstützt durch diesen bedingten Sensormechanismus kann das Modell aktive Stellen genau identifizieren und Enzymreste sowie funktionelle Substratgruppen untersuchen, die an wichtigen katalytischen Wechselwirkungen beteiligt sind, und zwar auf effiziente und kostengünstige Weise, ohne dass Strukturinformationen erforderlich sind.
Mithilfe künstlicher Intelligenz wird das Team die eingehende Forschung in verwandten Bereichen der KI für die Biowissenschaften weiter vorantreiben und mehr Möglichkeiten für die Virusvorhersage, das Design von Proteinmedikamenten, die Impfstoffentwicklung usw. eröffnen. Wir freuen uns auf weitere Erfolge ihres Teams.
Quellen:
https://www.who.int/
https://news.pku.edu.cn/jxky/90d276ae5f8441849fd04372fd872154.htm
https://news.pkusz.edu.cn/info/1003/8711.htm









