Command Palette
Search for a command to run...
Professor Zheng Wei Von Der Nankai-Universität: AlphaFold Ist Nicht Perfekt Und Die Akademische Gemeinschaft Hat Immer Noch Die Möglichkeit, „auf Der Kurve Zu überholen“.

In den letzten Jahren hat sich das Feld der Proteinstrukturvorhersage mithilfe von KI-Technologien wie Deep Learning rasant entwickelt. Im Oktober 2024 erhielten Demis Hassabis und John M. Jumper von DeepMind für AlphaFold den Nobelpreis für Chemie 2024. Dies bedeutet jedoch nicht, dass AlphaFold unersetzlich ist und dass es sich weiterhin lohnt, andere hervorragende Algorithmen zu erkunden.
In der sechsten Folge der Live-Serie „Meet AI4S“HyperAI fühlt sich geehrt, Professor Zheng Wei, Professor an der School of Statistics and Data Science der Nankai University, eingeladen zu haben.Unter dem Motto „Der Thron von AlphaFold3 ist nicht stabil und die akademische Gemeinschaft überholt ihn: Dreidimensionale Strukturvorhersage biologischer Makromoleküle und ihrer Wechselwirkungen auf der Grundlage von Deep Learning“ teilte er allen die Einschränkungen von AlphaFold und zukünftige Optimierungsrichtungen mit und erklärte, welche Algorithmen und Forschungsthemen in der akademischen Gemeinschaft einer Erforschung wert sind.
* Folgen Sie dem offiziellen Konto und antworten Sie mit „Meet AI4S 6th“, um die Präsentations-PPT zu erhalten
HyperAI hat den ausführlichen Austausch organisiert und zusammengefasst, ohne die ursprüngliche Absicht zu verletzen. Nachfolgend finden Sie die Abschrift der Rede.
Einschränkungen von AlphaFold
Proteine sind die Grundpfeiler aller Lebensaktivitäten und die Vorhersage der dreidimensionalen Strukturen von Proteinen ist für das Verständnis biologischer Funktionen von entscheidender Bedeutung. Obwohl AlphaFold 2 von DeepMind die Vorhersage von Proteinstrukturen auf eine neue Ebene gebracht hat, bedeutet dies nicht, dass das End-to-End-Framework von AlphaFold 2 alle Probleme der Proteinstrukturvorhersage gelöst hat.
Zunächst einmal weist AlphaFold 2 selbst am Beispiel noch viele Einschränkungen auf:
* Die Genauigkeit muss verbessert werden
Offizielle Berichte zeigen, dass AlphaFold 2 Strukturen mit einer Genauigkeit von mehr als 90% vorhersagen kann, die eigentliche Aufgabe kann jedoch kein so hohes Niveau erreichen.
* Die Vorhersage der Proteinstruktur mehrerer Domänen ist begrenzt
AlphaFold 2 liefert gute Ergebnisse bei der Vorhersage von Proteinen mit einer einzigen Domäne, bei komplexen Proteinen mit mehreren Domänen, bei denen die Domänen relativ flexibel sind, ist die Vorhersagegenauigkeit jedoch nicht gut.
* Die Vorhersage der Proteinkomplexstruktur ist begrenzt
Um zu funktionieren, müssen Proteine normalerweise Komplexe mit anderen Proteinen bilden, doch die erste Version von AlphaFold 2 ging auf dieses Problem nicht ein.
* Die Vorhersage von RNA-Strukturen, RNA-RNA- und Protein-RNA-Strukturen ist begrenzt
Wie oben erwähnt, wurden diese Probleme in der ursprünglichen Version nicht behoben.
* Die Vorhersage der Proteindynamik/Konformationsänderungen ist begrenzt
Experimentelle Analysemethoden können normalerweise nur den Strukturzustand zu einem bestimmten Zeitpunkt erfassen, Proteine existieren jedoch nicht statisch in ihren Organismen und ihre Strukturen können zu verschiedenen Zeitpunkten unterschiedlich sein. Diese Probleme wurden von AlphaFold 2 noch nicht gelöst.
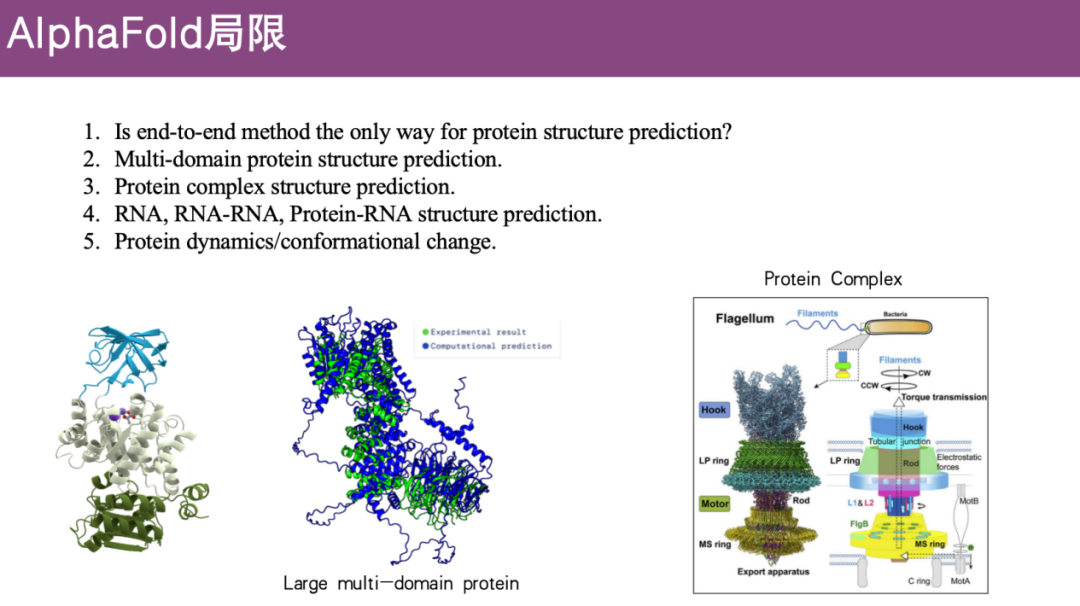
Und obwohl DeepMind AlphaFold 3 iteriert hat und wir alle wissen, dass es bei der Vorhersage von Proteinmonomerstrukturen gute Ergebnisse liefert, muss seine Genauigkeit bei der Vorhersage von Komplexen, Nukleinsäuren und kleinen Molekülen noch verbessert werden. daher,Die nächste Generation von AlphaFold könnte Vorhersagemodule mit anderen Funktionen hinzufügen.Da die vorhandenen Modelle hauptsächlich zur Behandlung statischer Strukturen verwendet werden, werden wir beispielsweise molekulardynamische Prozesse untersuchen und Proteinkonformationen vorhersagen. Darüber hinaus könnte es auch den Bereich des Proteindesigns betreffen und den gesamten Vorhersageprozess umkehren.
Daher bleibt auch mit AlphaFold im gesamten akademischen Bereich noch viel zu tun.
Gibt es neben AlphaFold noch andere Methoden, die es wert sind, erkundet zu werden?
In der Vergangenheit waren die wichtigsten Methoden, die wir zur Aufklärung der dreidimensionalen Struktur von Proteinen verwendeten, Röntgenstrahlen, Kernspinresonanz (NMR) und Kryo-Elektronenmikroskopie. Da die experimentelle Aufklärung von Proteinstrukturen schwierig und teuer ist, müssen manche Teams möglicherweise Monate oder sogar Jahre damit verbringen, die dreidimensionale Struktur eines Proteins aufzuklären. Infolgedessen begann man, eine kostengünstigere und schnellere Methode zu erforschen, nämlich die Vorhersage der Proteinstruktur mithilfe von Algorithmen.
Wir wissen, dass Proteine hauptsächlich aus 20 Arten von Aminosäuren bestehen, die normalerweise durch englische Buchstaben dargestellt werden, und dass Aminosäuremoleküle auch viele Atome enthalten.Daher kann das Problem der Vorhersage einer Proteinstruktur wie folgt zusammengefasst werden: Geben Sie eine Aminosäurezeichenfolge ein, die aus diesen Buchstaben besteht, und verwenden Sie einen Rechenalgorithmus, um die dreidimensionalen räumlichen Koordinaten (x, y, z) jedes Atoms in jeder Aminosäure in der Proteinsequenz vorherzusagen.
Betrachtet man die gesamte Entwicklungsgeschichte der Proteinstrukturvorhersage, so sind in unterschiedlichen Stadien eine Vielzahl repräsentativer Algorithmen entstanden, wie etwa vergleichende Modellierung oder Homologiemodellierung, Molekulardynamiksimulation (MD), Threading-Algorithmus, De-novo-Vorhersage und Strukturvorhersagealgorithmus basierend auf der Deep-Learning-Vorhersage von Kontaktkarten. Die Haupteinführung lautet wie folgt:
* Vergleichende Modellierung oder Homologiemodellierung
Diese Methode basiert auf den Prinzipien der biologischen Evolution.Man geht davon aus, dass bei hoher Sequenzähnlichkeit auch die Struktur und Funktion des Proteins relativ ähnlich sind.Daher können wir zunächst die Aminosäuresequenz des unbekannten Proteins ermitteln und dann durch Sequenzausrichtung die aufgelöste Proteinstrukturvorlage mit hoher Sequenzähnlichkeit in der PDB-Datenbank finden und durch Migration oder Ausrichtung die Struktur des unbekannten Proteins vorhersagen.
*Die PDB-Datenbank enthält die Strukturen von Proteinen, die in diesem Bereich gelöst wurden

* Molekulardynamische Simulation
Die Grundidee besteht darin, auf Grundlage der Aminosäuresequenz des Proteins zufällig eine anfängliche dreidimensionale Struktur zu erzeugen, jedem Atom zufällige Koordinaten zuzuweisen, die Atomposition anzupassen und dann die Zustandsenergie des Proteins zu verschiedenen Zeitpunkten auf Grundlage des vorab konstruierten physikalischen Energiefelds zu berechnen.Die Struktur mit der niedrigsten Energie ist die vernünftige Proteinkonformation.

* Threading-Algorithmus
Ähnlich wie bei der Homologiemodellierung besteht der Unterschied darin, dass Proteine mit hoher Sequenzähnlichkeit zwar häufig eine ähnliche Struktur aufweisen, Proteine mit ähnlichen Strukturen jedoch eine geringe Sequenzähnlichkeit aufweisen können und für solche Proteine keine geeigneten Vorlageninformationen in der PDB-Datenbank gefunden werden können. Anschließend schlugen die Forscher das Konzept des Profils vor und verwendeten auf der Grundlage der gesammelten homologen Sequenzen die Methode der multiplen Sequenzalignmentierung (MSA), um verschiedene Aminosäuren auf dieselbe Weise auszurichten wie zwei Proteinprofile.
Das heißt, selbst wenn die beiden Aminosäuresequenzen unterschiedlich sind,Da ihre Profile jedoch ähnlich sind, können wir davon ausgehen, dass auch ihre Strukturen ähnlich sind.Verwenden Sie dies, um Vorlagen zu finden.

* De-novo-Vorhersage
Für manche Proteine sind die Strukturen in der Datenbank möglicherweise nicht ähnlich.Anschließend versuchten die Forscher, Vorhersagen zu treffen, indem sie die gesamte Proteinsequenz in kürzere Fragmente zerlegten, in der Datenbank nach Vorlagen dieser kleinen Fragmente suchten und diese kleinen Fragmentvorlagen dann zu einer vollständigen dreidimensionalen Struktur zusammensetzten.
Insbesondere hat Professor David Baker von der University of Washington die Software Rosetta entwickelt, deren Hauptprinzip darin besteht, die Proteinsequenz in viele kleine Fragmente zu zerlegen, diese Fragmente zufällig zusammenzusetzen und sie dann mithilfe der in der molekularen Dynamiksimulation entwickelten Energiefunktion zu optimieren und eine Strukturvorhersage durch ähnliche Prinzipien wie die dynamische Simulation und Energieminimierung durchzuführen.
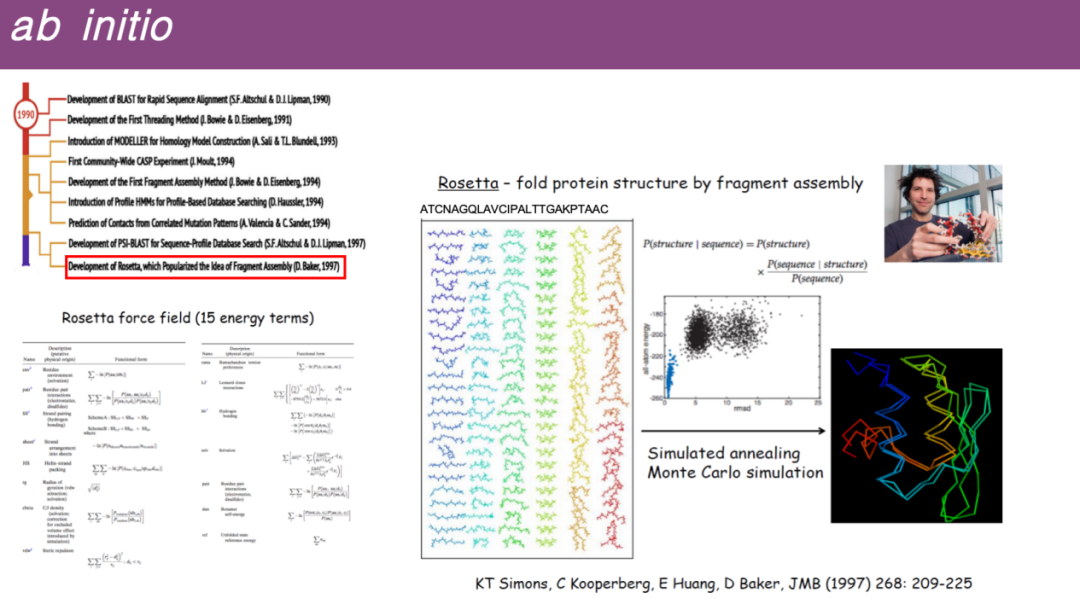
* Kontaktkarte
Die Grundidee besteht darin, die dreidimensionale Struktur eines Proteins in ein zweidimensionales Diagramm umzuwandeln.Dabei werden die dreidimensionalen Strukturinformationen des Proteins genutzt, d. h. die Koordinatenpositionen aller Punkte im Raum werden verwendet, um den Abstand zwischen verschiedenen Aminosäuren zu berechnen. Es wird angenommen, dass ein Kontakt entsteht, wenn der Abstand zwischen zwei Aminosäuren kleiner als ein bestimmter Schwellenwert ist, andernfalls kommt kein Kontakt zustande. Diese Definition wird verwendet, um die dreidimensionale Struktur in einen zweidimensionalen Graphen zu komprimieren. Darüber hinaus können die Informationen dieser zweidimensionalen Kontaktkarte verwendet werden, um die dreidimensionale Struktur des Proteins zu rekonstruieren.
Insbesondere haben die Forscher viele auf Deep Learning basierende Methoden entwickelt. Die Kernidee besteht darin, zunächst eine multiple Sequenzalignment (MSA) zu erstellen, um die koevolutionären Informationen der Profile in den Aminosäuren i und j zu beobachten, da solche koevolutionären Aminosäuren häufig räumlich sehr nahe beieinander liegen und Kontakte bilden. Anschließend werden die Informationen zur Koevolution als Features in das Deep-Learning-Netzwerk eingegeben, um es zu trainieren. Dadurch wird die Proteinkontaktkarte vorhergesagt und die dreidimensionale Struktur des Proteins wiederhergestellt.
Beispielsweise hat das Team von Professor Zheng Wei zuvor einen Algorithmus namens CI-TASSER entwickelt, der derzeit eine häufig verwendete Methode zur Vorhersage der Proteinstruktur auf der Grundlage von Kontaktkarten ist.
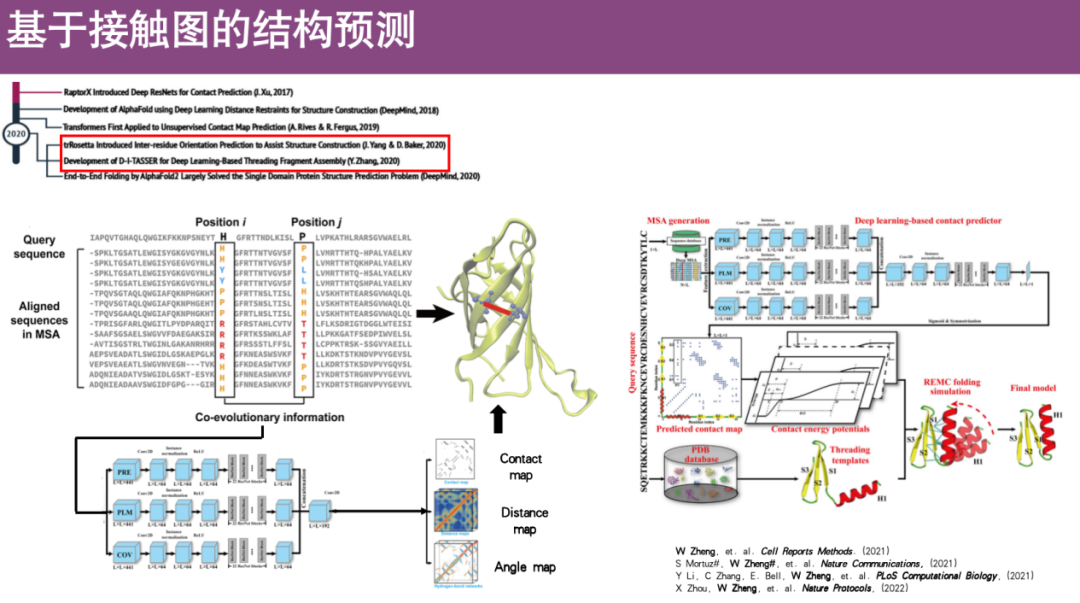
Schließlich integriert AlphaFold die Grundprinzipien vieler der oben genannten Algorithmen und erstellt erfolgreich ein End-to-End-Framework, das Proteinsequenzen direkt eingeben und dann Strukturen ausgeben kann.
Am Beispiel der Teamleistungen werden die Möglichkeiten für Akademiker untersucht,
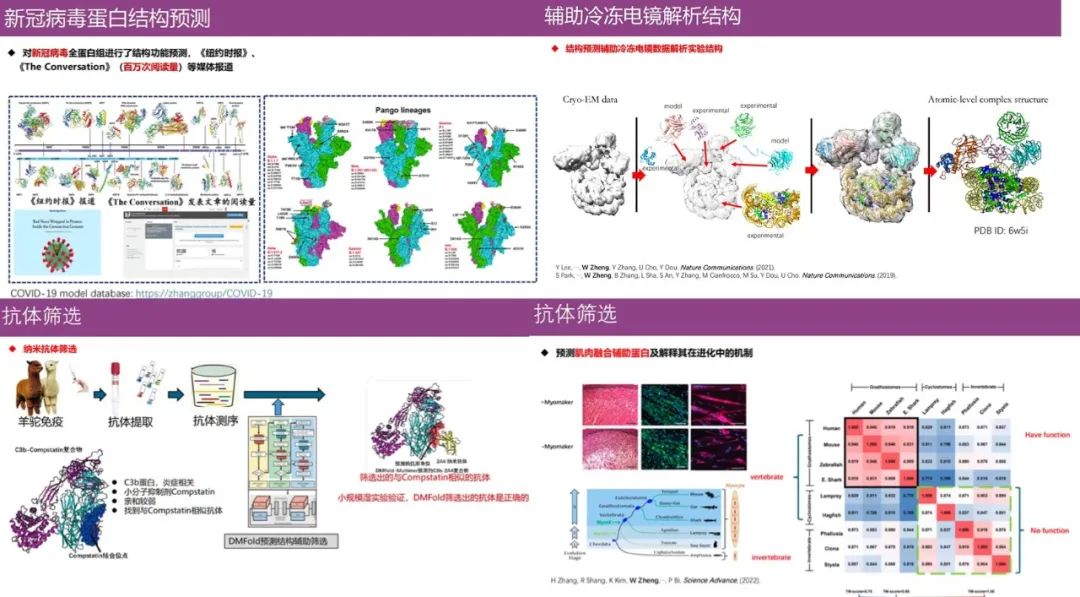
Die Vorhersage von Proteinstrukturen hat einen enormen Einfluss auf den biomedizinischen Bereich, zum BeispielDie Algorithmen, die derzeit vom Team um Professor Zheng Wei entwickelt werden, dienen der Vorhersage unbekannter viraler Proteinstrukturen (neues Coronavirus), der Unterstützung bei der Analyse von Proteinstrukturen mittels Kryo-Elektronenmikroskopie, der Unterstützung von Biologen beim Verständnis der evolutionären Funktionen von Proteinen sowie dem Antikörper-Screening.
Darüber hinaus wurden, wie in der Abbildung unten gezeigt, alle vom Team entwickelten Algorithmen zur Vorhersage von Proteinmonomeren und komplexen Strukturen in automatische Serveralgorithmen umgewandelt und auf der Website der Forschungsgruppe veröffentlicht. Seine Algorithmen haben bereits mehr als 90.000 Benutzern in über 100 Ländern auf der ganzen Welt geholfen und jeder ist herzlich eingeladen, sie zu verwenden.
*Gesamtprojektadresse:
https://seq2fun.dcmb.med.umich.edu/DMFold

Methode zur Vorhersage der Proteinmonomerstruktur DI-TASSER
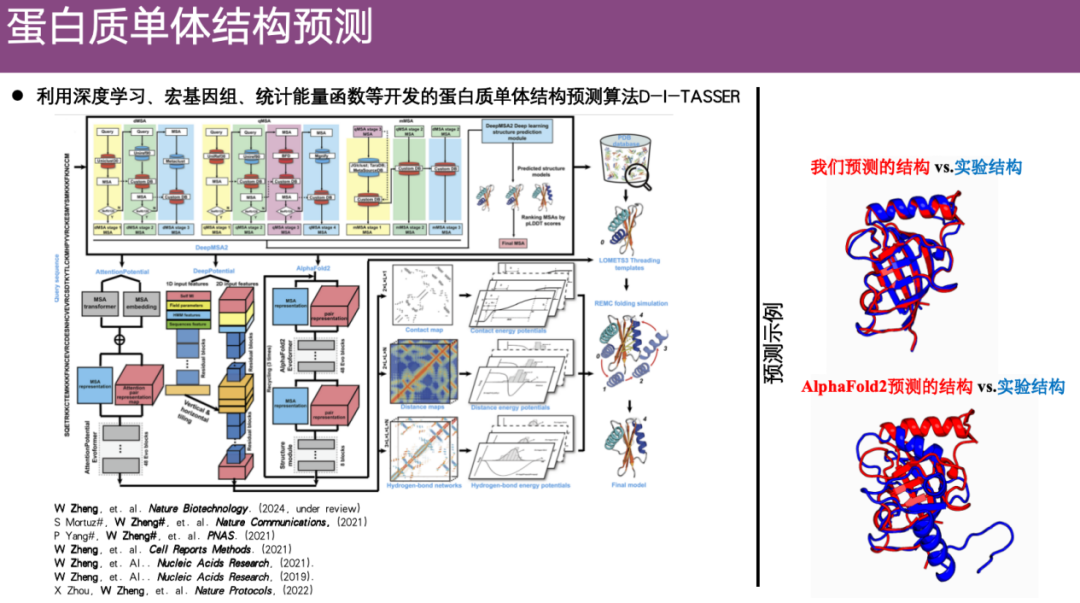
Das Problem der Vorhersage der Struktur von Proteinmonomeren hat schon immer viel Aufmerksamkeit auf sich gezogen. Vor AlphaFold 2 hatte das Team von Professor Zheng Wei Strukturvorhersageforschung auf der Grundlage von Kontaktkarten durchgeführt. Nach dem Erscheinen von AlphaFold 2 fragte sich das Team, ob es räumliche Einschränkungen wie die von AlphaFold 2 vorhergesagte Kontaktkarte in die zuvor entwickelten Algorithmen integrieren könnte. Daher basierend auf räumlichen Einschränkungen, Metagenomen, statistischen Energiefunktionen usw.Das Team entwickelte einen Algorithmus zur Vorhersage der Proteinmonomerstruktur DI-TASSER, der nach der Optimierung gute Ergebnisse zeigte.
Wie im Beispiel auf der rechten Seite der Abbildung unten gezeigt, stellt Rot die von DI-TASSER vorhergesagte Proteinstruktur dar und Blau die experimentell analysierte Struktur. Wie Sie sehen können,Die von DI-TASSER vorhergesagte Struktur ist der experimentell aufgelösten Struktur sehr ähnlich.Im Gegensatz dazu unterscheidet sich die von AlphaFold 2 vorhergesagte Struktur auch nach der Ausrichtung erheblich von der experimentellen Struktur und ihre Vorhersagegenauigkeit ist etwas geringer.
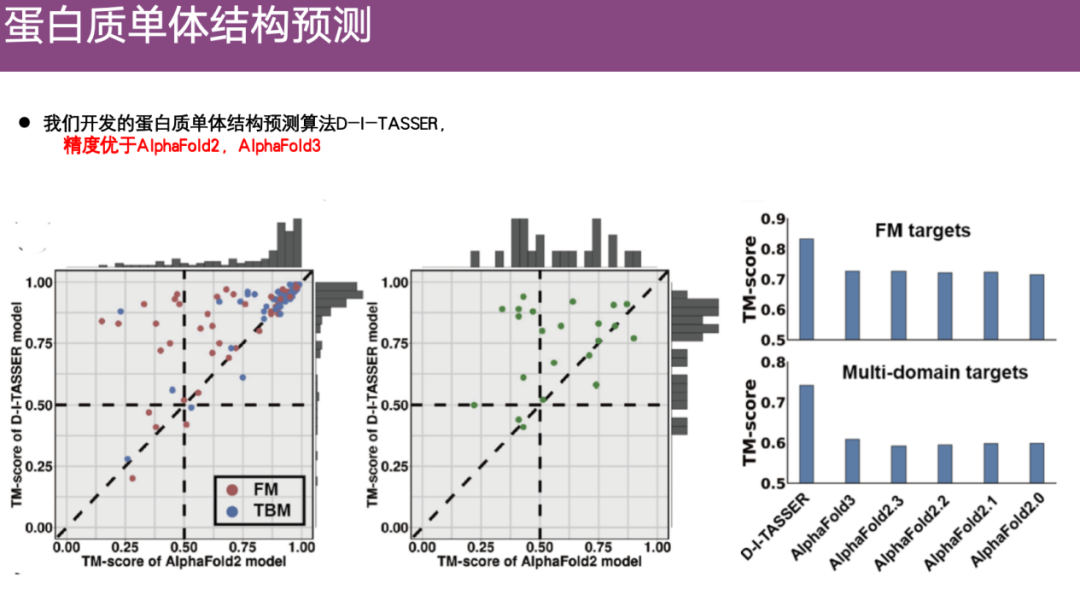
Darüber hinaus wird es anhand mehrerer Protein-Datensätze ausgewertet. Wie in der rechten Seite der Abbildung unten gezeigt, gilt bei der Vorhersage von Einzeldomänen und Multidomänen:Die Vorhersagegenauigkeit von DI-TASSER ist höher als die von AlphaFold 2 und sogar höher als die von AlphaFold 3.
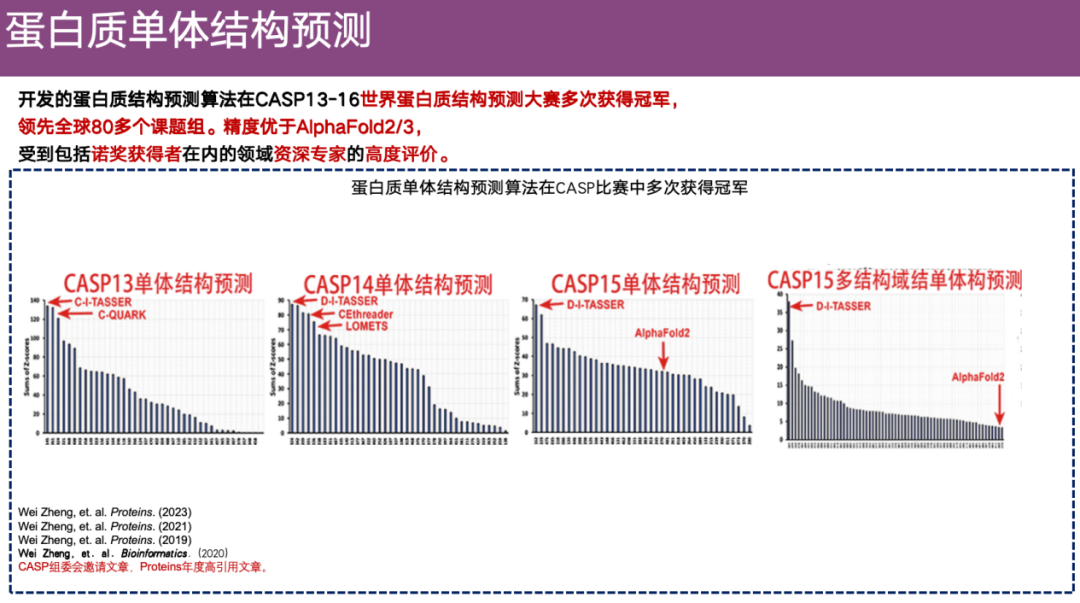
Um die Autorität der Bewertung sicherzustellen, führte das Team nicht nur interne Bewertungen durch, sondern nahm auch am maßgeblichen Wettbewerb auf diesem Gebiet teil – CASP.
Der CASP-Wettbewerb gilt als Olympiade der Branche und zielt in erster Linie darauf ab, Bewertungsmethoden für die Vorhersage von Proteinstrukturen zu standardisieren. Da es viele Arten von Algorithmen zur Vorhersage der dreidimensionalen Struktur von Proteinen gibt, hat jedes Labor auch seinen eigenen Algorithmus entwickelt. Da die Auswertungsdatensätze und -methoden unterschiedlich sein können, behauptet jede Forschungsgruppe normalerweise, dass ihre Methode die genaueste der Welt sei. Um dieses Durcheinander zu beseitigen, wurde der CASP-Wettbewerb ins Leben gerufen.
Seit letztem Jahr wurde CASP 16 Mal erfolgreich abgehalten und dauert nun schon 32 Jahre. Viele namhafte Teams nahmen daran teil, beispielsweise das Team von Professor David Baker und das Team von DeepMind.
DI-TASSER und seine Vorgängeralgorithmen haben mehrfach an CASP-Wettbewerben teilgenommen. Während CASP 13-CASP 15 nahm diese Methode eine führende Position auf dem Gebiet der Vorhersage von Proteinmonomerstrukturen ein. In CASP 15,Der DI-TASSER-Algorithmus nahm auch an der Multidomänen-Bewertung teil und seine Gesamtgenauigkeit war besser als die aller teilnehmenden Forschungsgruppen.
DMFold, eine Methode zur Vorhersage der Struktur von Proteinkomplexen
Die größte Herausforderung bei der Vorhersage komplexer Strukturen besteht darin, die relative Torsion zwischen zwei Proteinen vorherzusagen, die mithilfe koevolutionärer Informationen analysiert werden kann.
Wenn man beispielsweise eine multiple Sequenzalignment (MSA) der Monomere zweier Proteine erstellt, die beiden MSAs anhand einiger Verbindungsmethoden zu einem MSA zusammenführt und die koevolutionäre Beziehung der Aminosäuren zwischen den beiden MSAs nutzt, um auf die Distanz zwischen Aminosäuren in unterschiedlichen Proteinen zu schließen, können die koevolutionären Informationen auch in das Deep-Learning-Framework integriert werden, um die relative Torsion zwischen zwei Proteinen vorherzusagen.
In diesem ZusammenhangDie Forschungsgruppe von Professor Zheng Wei entwickelte die Algorithmen DeepMSA und MetaSource, um tiefere multiple Sequenzalignments zu konstruieren.Darüber hinaus verwendete das Team Deep Learning, Metagenomik usw., um einen Algorithmus zur Vorhersage von Proteinkomplexstrukturen (DMFold) zu entwickeln.
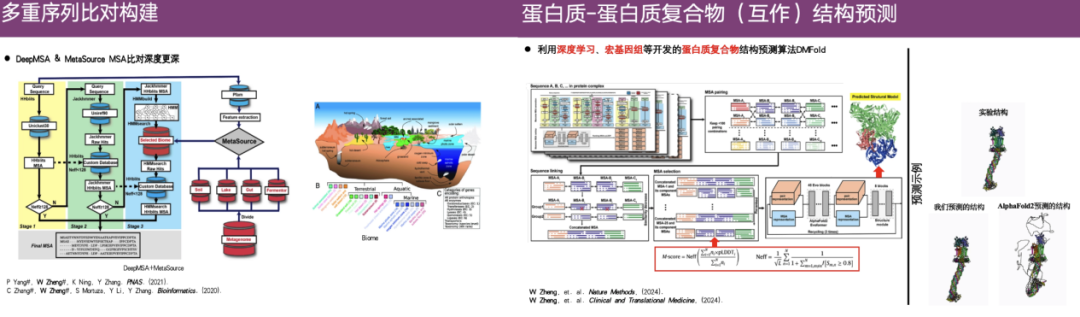
Wie im Fall ganz rechts in der obigen Abbildung gezeigt, ist oben die durch experimentelle Analyse erhaltene tatsächliche Struktur, unten links die von DMFold vorhergesagte Struktur und rechts das von AlphaFold 2 vorhergesagte Ergebnis. Es ist ersichtlich, dass die von AlphaFold 2 vorhergesagte Struktur relativ chaotisch ist und abnormale tentakelartige Verlängerungen aufweist. Im Gegensatz dazu weist die vorhergesagte Struktur von DMFold eine große Ähnlichkeit mit der experimentellen Struktur auf.Dies zeigt, dass der DMFold-Algorithmus AlphaFold 2 bei der Vorhersage komplexer Strukturen überlegen ist.
Darüber hinaus zeigt DMFold auch eine hohe Genauigkeit bei großen Protein-Protein-Komplexen, Nanoantikörper-Antigen-Komplexen, Konformationsänderungen durch Punktmutationen usw. Im CASP 15-Wettbewerb Das Gesamtranking von DMFold ist viel höher als das von AlphaFold 2 und in CASP 16 ist DMFold auch besser als AlphaFold 3.
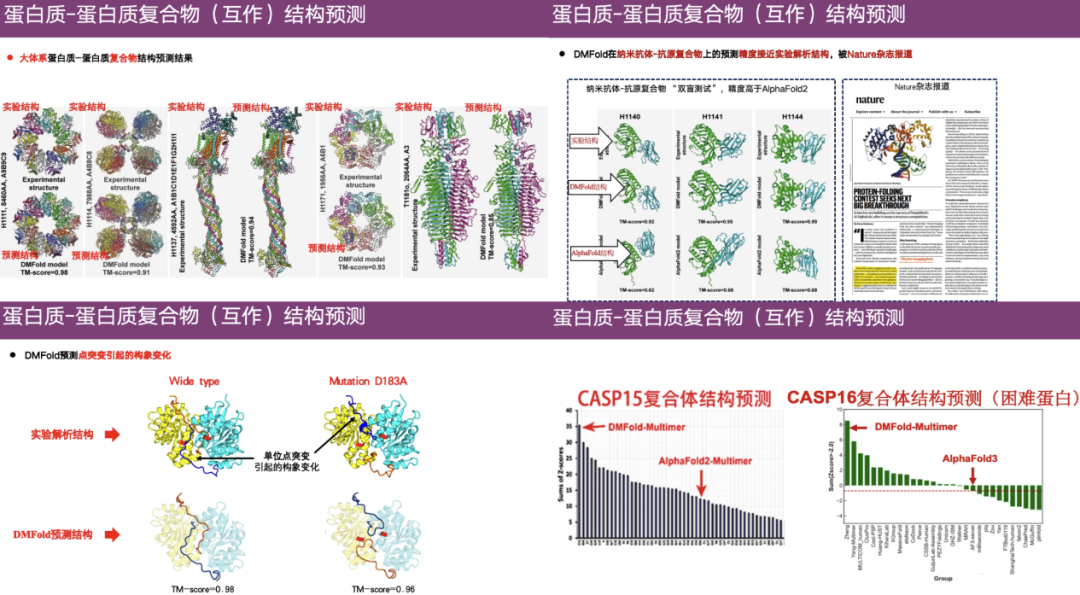
RNA-RNA-Komplexstrukturvorhersagemethode ExFold
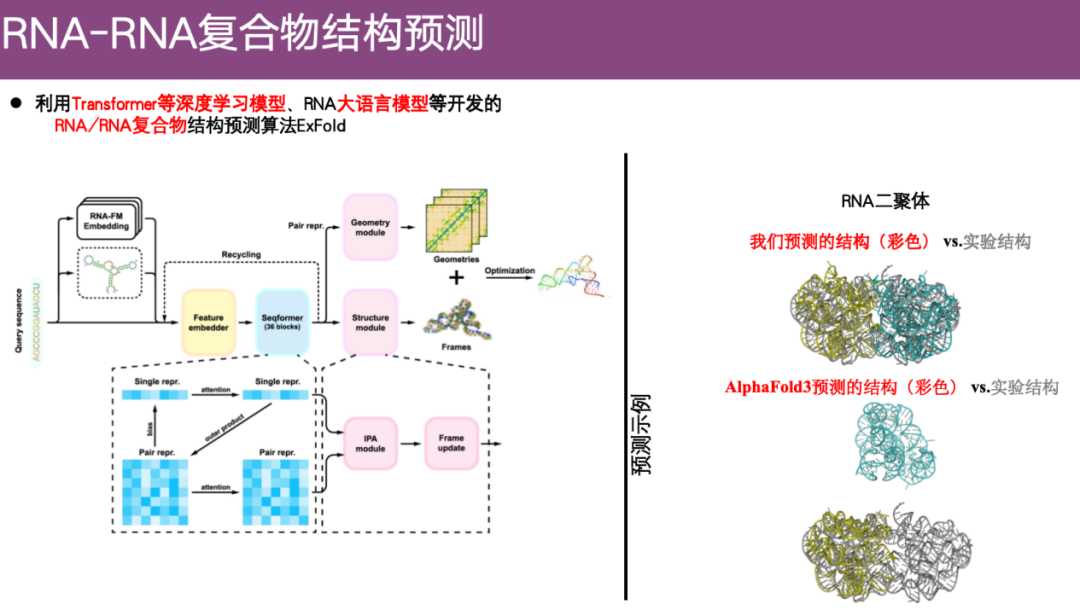
In den letzten Jahren hat das Team begonnen, sich auf das Problem der RNA-Strukturvorhersage zu konzentrieren. Beispielsweise haben sie den Algorithmus ExFold zur Vorhersage komplexer RNA/RNA-Strukturen unter Verwendung von Deep-Learning-Modellen wie Transformer und RNA-Big-Language-Modellen entwickelt.
Wie im Beispiel auf der rechten Seite der Abbildung unten gezeigt, ist der graue Teil die experimentelle Struktur und der farbige Teil die vorhergesagte Struktur. Wie Sie sehen können,Mithilfe der ExFold-Methode konnten die beiden Strukturen gut ausgerichtet werden. Im Gegensatz dazu zeigte die AlphaFold 3-Vorhersage, dass nicht einmal ein Kontakt zwischen den beiden RNA-Molekülen bestand, was fast als völlig falsch angesehen werden kann.
Das Team verglich auch die Genauigkeit von ExFold 3 mit AlphaFold 3 anhand eines größeren Datensatzes, wie auf der linken Seite der folgenden Abbildung dargestellt. Die Y-Achse stellt die Vorhersagegenauigkeit von ExFold dar.Die X-Achse stellt die Vorhersagegenauigkeit von AlphaFold 3 dar. Es ist ersichtlich, dass die Vorteile von ExFold immer noch ziemlich offensichtlich sind.
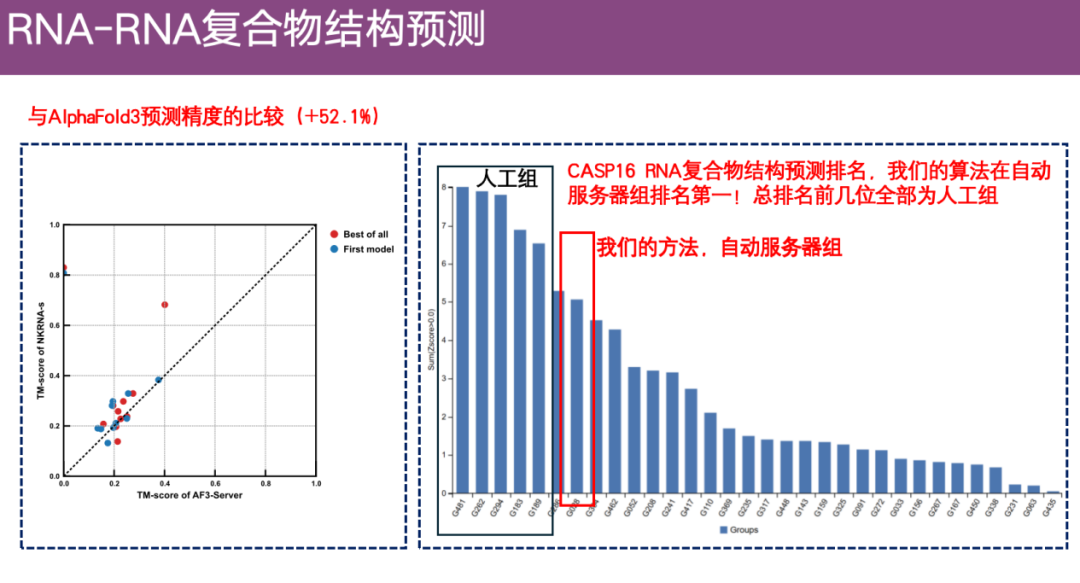
Darüber hinaus wurde im CASP 16-Wettbewerb zur Vorhersage der RNA-KomplexstrukturObwohl ExFold insgesamt nicht den ersten Platz belegt, ist es unter allen automatischen Algorithmen (Serveralgorithmen) der höchste.
* Der CASP-Wettbewerb ist in eine automatische und eine manuelle Gruppe unterteilt. Die automatische Gruppe muss die Vorhersageergebnisse innerhalb von 3 Tagen vollautomatisch übermitteln, und ein menschliches Eingreifen ist nicht zulässig. Die manuelle Gruppe hat 3 Wochen Zeit und kann Expertenerfahrungen und manuelle Anpassungen hinzufügen.
Methode zur Vorhersage der Protein-RNA-Komplexstruktur DeepProtNA
Im Hinblick auf das Problem der Vorhersage der Struktur von Protein-RNA-Komplexen verwendete das Team Deep-Learning-Modelle wie Transformer und das seit kurzem beliebte Large Language Model, um einen neuen Algorithmus zur Strukturvorhersage zu entwickeln: DeepProtNA.
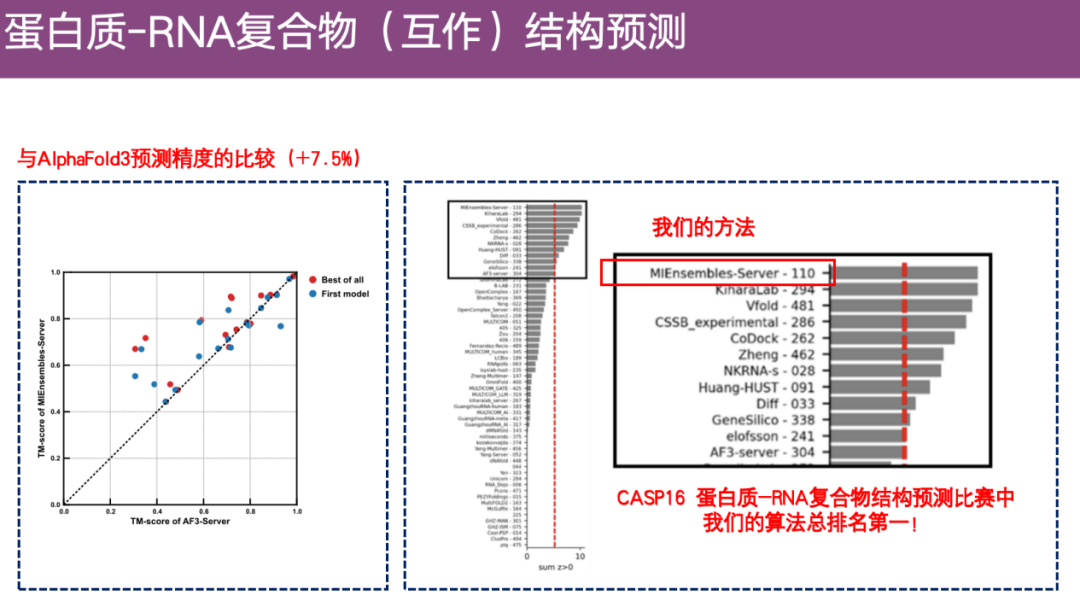
Wie im Beispiel rechts unten gezeigt, stellen im Antikörper-RNA-Komplex die Farben die Vorhersageergebnisse von DeepProtNA dar, während das Grau die experimentelle Struktur darstellt. Nach der Ausrichtung können wir feststellen, dassDie vorhergesagte Struktur von DeepProtNA stimmt weitgehend mit der experimentellen Struktur überein (Grau- und Farbüberlappung).Insbesondere an der Schnittstelle zwischen Antikörperprotein und Antigen-RNA ist die Vorhersagegenauigkeit sehr hoch. Im Gegensatz dazu überschneidet sich die vorhergesagte Struktur von AlphaFold 3 kaum mit der experimentellen Struktur und der Vorhersageeffekt ist gering.
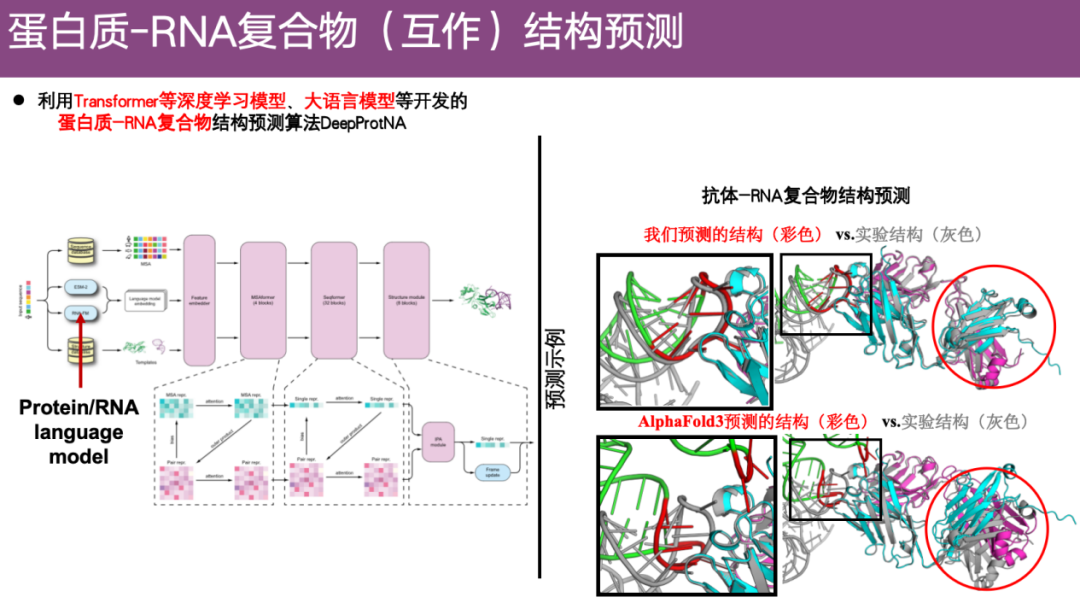
Auch,DeepProtNA ist etwa 7,5 Prozentpunkte genauer als AlphaFold 3.Erster Platz im Servergruppenwettbewerb von CASP 16.
EnsembleFold: Eine Methode zur Vorhersage allosterischer Strukturen von Biomakromolekülen
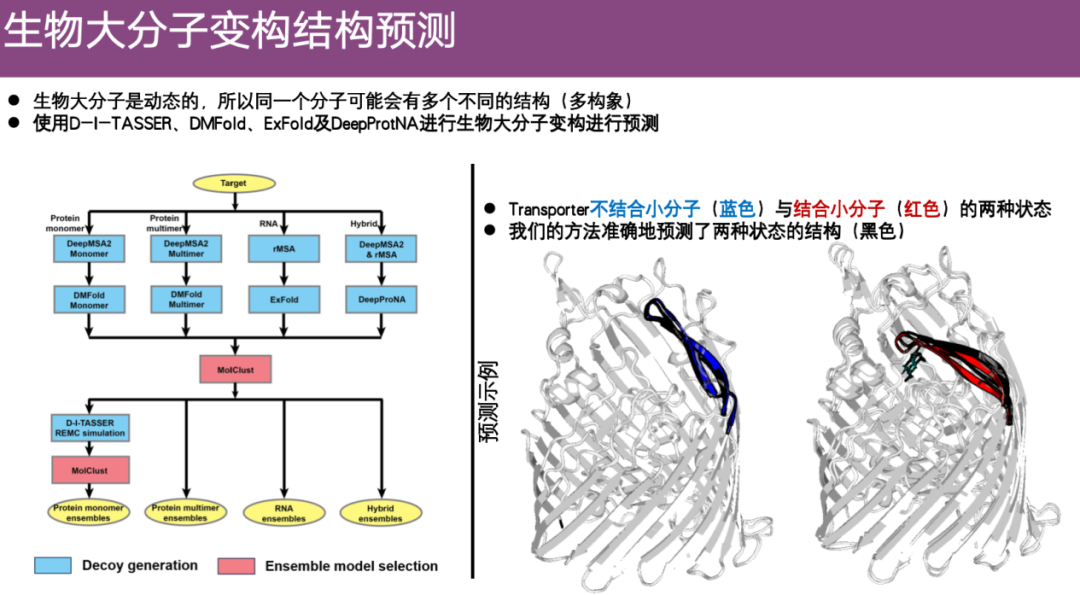
Das Team konzentriert sich außerdem auf das Problem der Vorhersage der allosterischen Struktur biologischer Makromoleküle. Die Eingabe des makromolekularen Multikonformationsproblems ist eine Proteinsequenz, und die Ausgabe sind mehrere Schlüsselbilder des Proteins in verschiedenen Zuständen. Dies bedeutet, dass im Vergleich zu statischen Vorhersagealgorithmen mehrere verschiedene Strukturen aus einer einzigen Aminosäuresequenz vorhergesagt werden müssen und diese Strukturen die Schlüsselrahmen des gesamten dynamischen Prozesses darstellen. Dies ist ein Thema, dem in der aktuellen Forschung große Aufmerksamkeit zuteilwird, dessen weitere Entwicklung jedoch schwer vorherzusagen ist.
Durch die Integration zuvor entwickelter Methoden und deren Optimierung für die makromolekulare AllosterieDas Forschungsteam entwickelte einige Clustering-Algorithmen und entwickelte schließlich einen Algorithmus namens EnsembleFold.
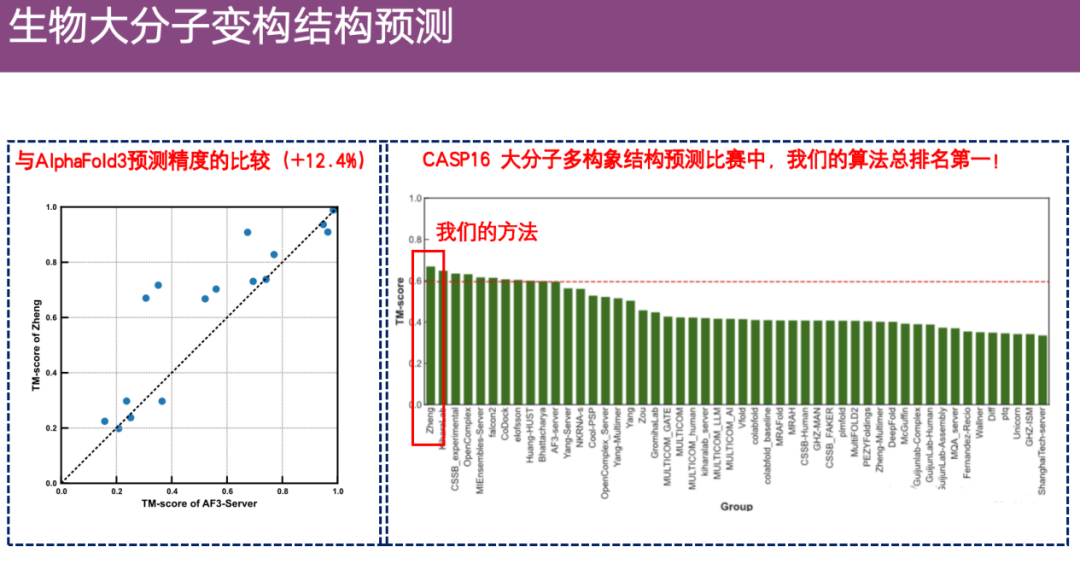
Wie im Beispiel auf der rechten Seite der Abbildung unten gezeigt, werden die Konformationsänderungen des Proteins nach der Bindung an das kleine Molekül demonstriert. Blau stellt die experimentelle Struktur dar, wenn das kleine Molekül nicht gebunden ist, und Rot stellt die Neigung und Konformationsänderung nach der Bindung an das grüne kleine Molekül dar. Das Team sagte auf Grundlage der eingegebenen Proteinsequenz zwei Strukturen voraus, die die schwarzen Teile darstellen. Es ist ersichtlich, dass die vorhergesagte Struktur von EnsembleFold sehr gut mit der tatsächlichen Struktur übereinstimmt, wenn es nicht an kleine Moleküle gebunden ist. Nach der Bindung an kleine Moleküle kann EnsembleFold auch die experimentelle Struktur gut anpassen. daher,EnsembleFold zeigt eine extrem hohe Genauigkeit bei der Vorhersage von Konformationsänderungen in Biomakromolekülen.
gleichzeitig,Nach einem Vergleich mit AlphaFold 3 stellte sich heraus, dass die Genauigkeit von EnsembleFold um etwa 12,4% höher war.Es belegt den ersten Platz unter allen makromolekularen Konformationswettbewerben in CASP 16.
Ein interessantes Beispiel ist die Vorhersage des Teams zu den Konformationsänderungen der Bakteriophagen-DNA-Integrase in CASP. Wie in der Abbildung unten gezeigt, wird die Aminosäuresequenz des Bakteriophagen durch P-P' dargestellt und die genetische Materialsequenz der Bakterien durch B-B'. Durch einen dynamischen Prozess integriert die Bakteriophagen-DNA-Integrase das genetische Material P' des Phagen in das genetische Material B der Bakterien, um B-P' zu bilden, und die Konformation ändert sich.
Das Team verwendete Algorithmen, um diese vielfältigen Konformationsänderungen vorherzusagen. Die experimentellen Strukturen sind links dargestellt, wobei sich oben der nicht integrierte Zustand (Konformation 1) und unten der integrierte Zustand (Konformation 2) befindet. Es ist ersichtlich, dass die Vorhersagen des Forschungsteams diese beiden unterschiedlichen Konformationen genau widerspiegeln können.
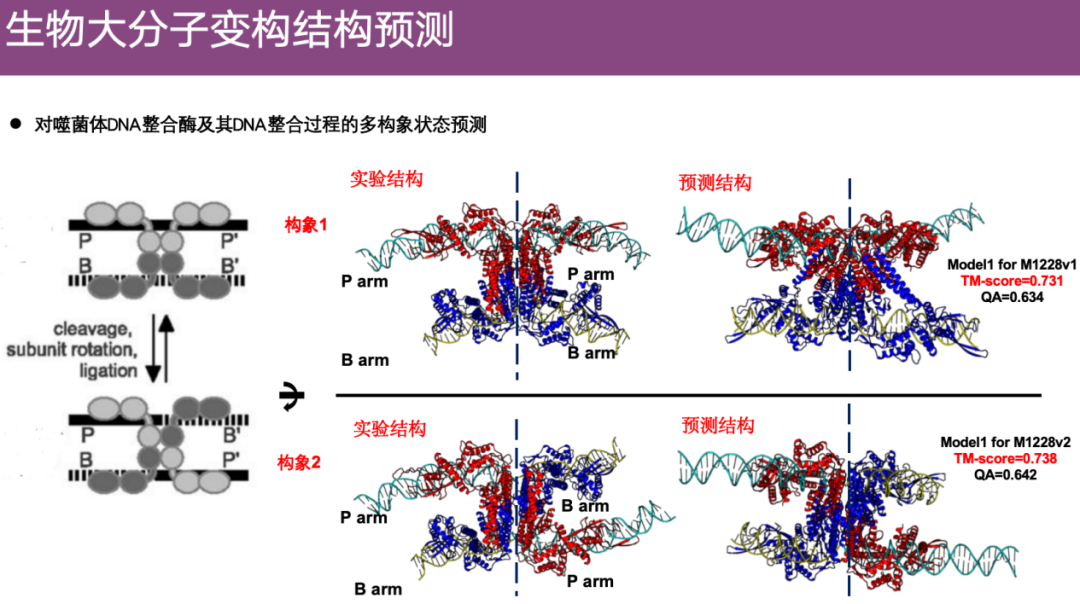
Es ist erwähnenswert, dass im CASP 16-WettbewerbDie Teilnehmer erhielten lediglich Sequenzinformationen und kannten weder den spezifischen biologischen Prozess noch Einzelheiten zu Konformationsänderungen. Dem Team von Professor Zheng Wei gelang es jedoch, den gesamten biologischen Prozess durch Vorhersage wiederherzustellen.Auch die Schiedsrichter zeigten sich in der Zusammenfassung nach dem Spiel überrascht.
Rekrutierung von Forschungsgruppen
Professor Zheng Wei von der School of Statistics and Data Science der Nankai University engagiert sich seit langem in der Vorhersageforschung zur Struktur, Funktion und Interaktion biologischer Makromoleküle wie Proteinen. Er leitete die Entwicklung einer Reihe von Algorithmen zur Strukturvorhersage und Strukturbewertung von Proteinmonomeren, Proteinkomplexen, Nukleinsäuren und Komplexen sowie Protein-Nukleinsäure-Komplexen mit besserer Genauigkeit als AlphaFold2/3. Er hat in vielen Wettbewerben der World Protein Structure Prediction Competition (CASP) (CASP13-16) die Meisterschaft gewonnen und leitet mehr als 80 akademische/industrielle Forschungsgruppen auf der ganzen Welt.
Das Bioinformatik-Team der School of Statistics and Data Science der Nankai-Universität, an der er arbeitet, sucht neue Mitglieder.Wenn Sie sich für computergestützte Strukturbiologie, Bioinformatik oder Datenwissenschaft interessieren, sind Sie – egal ob Master-, Doktor- oder Postdoktorand – herzlich willkommen, dem Team von Professor Zheng Wei beizutreten.
Interessierte Studierende können Professor Zheng Wei auf folgende Weise kontaktieren:
* E-Mail: [email protected]
* WeChat: 18622152765









