Command Palette
Search for a command to run...
Die Erfolgsrate Kann 100% erreichen. Das Arzneimittelentwicklungsunternehmen Cellarity Hat Sich Mit NVIDIA Zusammengetan, Um Zielgerichtete Moleküle Auf Basis Von Reinforcement Learning Zu Optimieren

Von der Antike bis zur Gegenwart hat die Menschheit nie aufgehört, gegen Krankheiten zu kämpfen. Die Entwicklung eines neuen Medikaments könnte Tausende von Leben retten und sogar die Lebenserwartung des Menschen insgesamt verlängern.
Wenn man auf die jahrhundertelange Geschichte der Arzneimittelforschung und -entwicklung zurückblickt, gibt es viele interessante Geschichten. So weichte beispielsweise im frühen 19. Jahrhundert der deutsche Apothekergehilfe Zeltina Opium in heißem Wasser ein und extrahierte es anschließend mit Ammoniakwasser, um einen Haufen weißes Pulver vom Opium zu trennen. Sie gab dem Hund dieses weiße Pulver und der Hund wurde kurz nach dem Fressen ohnmächtig.Deshalb nannte er es Morphin, nach dem griechischen Gott der Träume, Morpheus.Daher gilt Morphin allgemein als der weltweit erste aus einer Pflanze isolierte Wirkstoff und gilt auch als Ausgangspunkt moderner Arzneimittelinnovationen.
Anschließend beherrschten Apotheker nach und nach die Technologie zur Synthese chemischer Arzneimittel, und der deutsche Apotheker Selmann synthetisierte Acetylsalicylsäure, den Vorgänger von Aspirin. Zu Beginn des 20. JahrhundertsDie Nachfrage der Unternehmen nach neuen Medikamenten hat die Entwicklung der Hochdurchsatz-Screening-Technologie vorangetrieben, die es Wissenschaftlern ermöglicht, eine große Anzahl von Verbindungen effizienter zu screenen und zu testen. Zu Beginn des 21. JahrhundertsForscher haben begonnen, präzisere und wirksamere medikamentöse Behandlungen zu erforschen, wobei zielgerichtete Medikamente zu einer wichtigen Forschungsrichtung geworden sind.
Heute eröffnet die rasante Entwicklung der künstlichen Intelligenz neue Möglichkeiten für die Arzneimittelforschung. KI kann Apothekern dabei helfen, Wirkstoffziele schneller zu validieren und das Design von Arzneimittelstrukturen zu optimieren. Sie können sogar direkt Moleküle mit spezifischen physikochemischen Eigenschaften oder biologischen Aktivitäten erzeugen, was die Arzneimittelforschung erheblich beschleunigt.
In diesem ZusammenhangForscher des Biowissenschaftsunternehmens Cellarity und von NVIDIA haben gemeinsam eine neuartige gezielte molekulare Optimierungsmethode auf Basis des latenten Verstärkungslernens (MORL) vorgeschlagen.Der Ansatz kombiniert ein leistungsstarkes generatives Modell, das auf einem großen chemischen Datensatz vortrainiert wurde, mit einem hochmodernen Reinforcement-Learning-Algorithmus (RL) zur kontinuierlichen Raumoptimierung. Durch die Anwendung der Methode auf Aufgaben im Zusammenhang mit der Arzneimittelentdeckung, die Verwendung gängiger Benchmarks und den Vergleich mit modernsten Methoden stellten die Forscher fest, dass MOLRL bei einer Vielzahl von Aufgaben eine überlegene oder konkurrenzfähige Leistung zeigte, insbesondere bei der gezielten Molekülerzeugung und der Multiparameteroptimierung.
Die entsprechenden Ergebnisse wurden auf ChemRxiv unter dem Titel „Targeted Molecular Generation With Latent Reinforcement Learning“ veröffentlicht.
Papieradresse:
Folgen Sie dem offiziellen Konto und antworten Sie mit „Zielmoleküloptimierung“, um das vollständige PDF zu erhalten
Das Open-Source-Projekt „awesome-ai4s“ vereint mehr als 100 AI4S-Papierinterpretationen und stellt umfangreiche Datensätze und Tools bereit:
https://github.com/hyperai/awesome-ai4s
Routenauswahl: direkte Modifizierung von Molekülen vs. Arbeiten im latenten Raum
Die Arzneimittelentwicklung ist ein sehr komplexer Prozess – zusätzlich zur biologischen Aktivität muss eine Verbindung über zahlreiche weitere Eigenschaften verfügen, um als klinischer Kandidat ausgewählt zu werden. Die Strukturen der Verbindungen, denen eine therapeutische Wirksamkeit zugeschrieben wird (die oft als „Kandidatenverbindungen“ bezeichnet werden), sind nicht in Stein gemeißelt, sondern werden über einen langen iterativen Zyklus hinweg modifiziert, um Probleme wie unzureichende Löslichkeit und Wirksamkeit zu beheben.
In einem iterativen Prozess transformieren Pharmazeuten typischerweise Ausgangsmoleküle, um Analoga zu entwerfen, die auf ihrer Intuition oder durch Aufzählung aus reaktionsbasierten Bibliotheken basieren. Angesichts der enormen Größe des chemischen Raums gestaltet sich das Design jedoch selbst für ein einzelnes Molekül äußerst schwierig und erfordert eine umfassende Auswertung des gesamten chemischen Raums. Computergestützte Methoden zur gezielten Molekülerzeugung können den chemischen Raum effizient erkunden und Chemikern bisher unerforschte Strukturen empfehlen.
Derzeit können Methoden zur Zielmolekülgenerierung und -optimierung in zwei Kategorien unterteilt werden:Die erste Methode besteht darin, direkt auf die Molekülstruktur einzuwirken.Strukturelle Modifikationen zu identifizieren, die die Zieleigenschaften verbessern;Die zweite Kategorie von Methoden operiert im latenten Raum des generativen Modells.Verändern Sie die Molekülstruktur indirekt über ihre latente Darstellung.
Mit Methode 1 können Strukturänderungen durch Einfügen oder Entfernen von Atomen oder chemischen Bindungen vorgenommen werden, und die Industrie hat dabei erhebliche Fortschritte gemacht.
Es wurde berichtet, dass im November letzten Jahres ein Team unter der Leitung von Professor Yoonsu Park vom Korea Advanced Institute of Science and Technology (KAIST) eine innovative Technologie zur Einzelatom-Bearbeitung entwickelt hat. Diese Technologie führt Photokatalysatoren ein.Die Einzelatombearbeitung von Arzneimittelmolekülen wurde bei Raumtemperatur und -druck erfolgreich durchgeführt.Die vom Team entwickelte „Molekularscheren“-Technologie kann Fünfringstrukturen präzise schneiden und verbinden, wobei Sauerstoffatome durch Stickstoffatome ersetzt werden, wodurch die molekularen Eigenschaften verändert und die Wirksamkeit des Medikaments verbessert wird. Die entsprechenden Forschungsergebnisse wurden in Science unter dem Titel „Photocatalytic furan-to-pyrrole conversion“ veröffentlicht.

Allerdings ist es nicht einfach, nach Belieben Operationen an Molekülen durchzuführen. Einerseits können Strukturveränderungen gegen die Regeln der Chemie verstoßen und so zu ungültigen Molekülstrukturen führen. Da Molekülstrukturen andererseits von Natur aus diskret sind und das Hinzufügen oder Entfernen chemischer Bindungen diskrete Vorgänge erfordert, führt diese Diskretheit zu diskontinuierlichen Gradienten im Optimierungsprozess, was die effektive Anwendung gradientenbasierter Methoden erschwert.
Im Vergleich zu Methode 1Der zweite Ansatz wandelt die Optimierungsaufgabe in ein kontinuierliches Optimierungsproblem um, nutzt den latenten Raum des generativen Modells und verwendet kontinuierliche Raumoptimierungsalgorithmen wie den Gradientenabstieg.Dennoch bleibt die chemische Validität eine Herausforderung, da es keine Garantie dafür gibt, dass ein Punkt im latenten Raum einem gültigen Molekül entspricht. Durch die Verwendung neuartiger Architekturen und Trainingsmodifikationen konnten bei generativen Modellen jedoch erhebliche Fortschritte bei der Verbesserung der Effektivität und Kontinuität im latenten Raum erzielt werden.
In der Forschung von Cellarity und NVIDIA schlugen die Forscher MOLRL vor, um im latenten Raum des vortrainierten generativen Modells mithilfe der Methode der proximalen Policy-Optimierung (PPO) zu optimieren.
MOLRL, eine gezielte molekulare Optimierungsmethode basierend auf latentem Verstärkungslernen
Wie funktioniert das MOLRL-Framework?
Das MOLRL-Framework besteht aus zwei Teilen: einem generativen Latent-Space-Modell und einem Reinforcement-Learning-Agenten (RL).
Das generative Modell ist ein vortrainiertes Encoder-Decoder-Modell, dessen latenter Raum den chemischen Raum kodiert, über den der RL-Agent operiert. Der RL-Agent wird mit der PPO-Methode trainiert,Im latenten Raum navigieren; Die Belohnungsfunktion gibt dem Agenten Feedback,Helfen Sie ihnen, sich im Raum zurechtzufinden,Identifizieren Sie Moleküle mit gewünschten Eigenschaften.
Wie unten gezeigt: Die latente Darstellung „z“ des Eingabemoleküls wird durch die Aktion „a“ gestört, die aus der Ausgabe des Richtliniennetzwerks extrahiert wird. Der gestörte latente Vektor „z‘“ wird in Moleküle dekodiert und durch eine Belohnungsfunktion bewertet. Der Status „z“, die Aktion „a“ und die Belohnung „R“ werden erfasst und zum Aktualisieren des Richtliniennetzwerks verwendet.
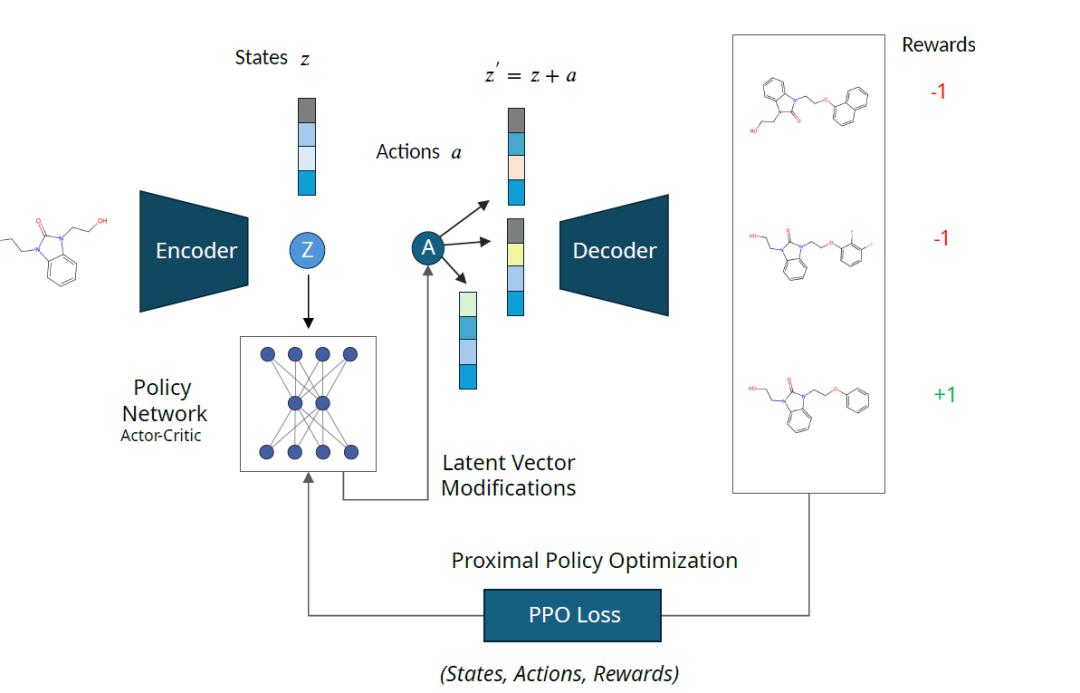
Das Framework ist unabhängig von der Architektur des Encoders und Decoders, die Eigenschaften des latenten Raums wirken sich jedoch stark auf die Optimierungsleistung aus. Daher bewerteten die Forscher die Leistung von MOLRL auf zwei verschiedenen Encoder-Decoder-Architekturen, nämlich dem Variational Autoencoder (VAE) und dem auf der Grundlage von Mutual Information Machine Learning (MolMIM) trainierten Autoencoder.
Ein Reinforcement-Learning-Agent (RL) ist für die Navigation im latenten Raum verantwortlich, um Moleküle mit gewünschten molekularen Eigenschaften zu identifizieren. Die Forscher verwendeten PPO oder Proximal Policy Optimization, um den RL-Agenten zu trainieren.Der PPO-Algorithmus führt den Agenten zum optimalen Pfad im latenten Raum, indem er die Richtlinie optimiert, um die langfristige kumulative Belohnung zu maximieren.Die Belohnungsfunktion ist der Kern des MOLRL-Frameworks, das dem Agenten Feedback basierend auf den Zieleigenschaften des Moleküls (wie Arzneimittelähnlichkeit, synthetische Zugänglichkeit, Zielbindung usw.) liefert.
Wie ist die Leistung des MOLRL-Frameworks?
Um die Leistung des MOLRL-Frameworks zu bewerten, entwickelten die Forscher eine mehrzielige Optimierungsaufgabe und verglichen sie mit den aktuellen modernsten Optimierungsmethoden.
Konkret wandten die Forscher MOLRL an, um biologisch aktive Moleküle zu erzeugen, die auf zwei Ziele abzielen, und dabei sowohl die Arzneimittelähnlichkeit (QED) als auch die synthetische Zugänglichkeit (SA) zu optimieren. Als biologische Ziele wurden zwei mit der Alzheimer-Krankheit assoziierte Kinasen ausgewählt – GSK3β und JNK3. Basierend auf der Bewertungsstrategie von Jin et al. zeichneten die Forscher die 5.000 Moleküle mit den höchsten Belohnungswerten auf, die während des Optimierungsprozesses generiert wurden, und berechneten die folgenden drei Indikatoren: Erfolgsrate; Neuheit; und Vielfalt.
Die folgende Tabelle zeigt die Leistung von MOLRL, das im latenten VAE-CYC-Raum trainiert wurde, und MOLRL, das im MolMIM-Raum trainiert wurde, sowie den Leistungsvergleich der aktuellen hochmodernen molekularen Optimierungsmethoden, über die in der Literatur berichtet wird.
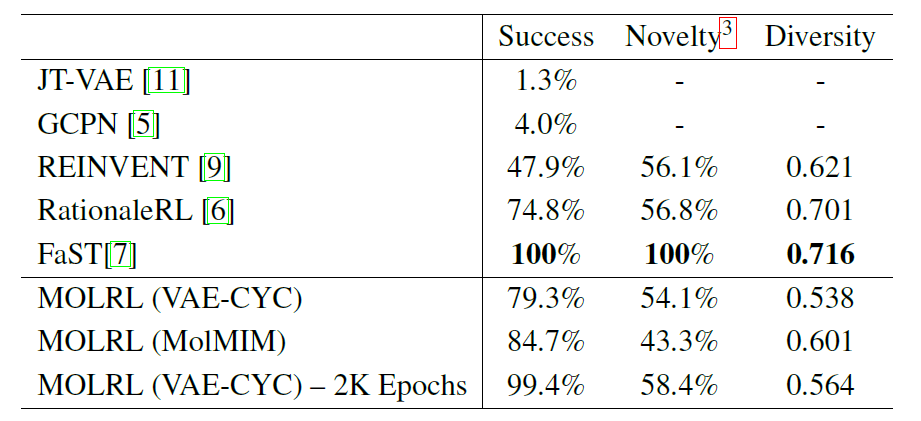
Wie in der Tabelle gezeigt, erstellt FaST Molekülgraphen durch die Kombination Molekülfragmente mithilfe von Reinforcement Learning (RL).Es zeigt eine höhere Erfolgsquote unter allen verglichenen Methoden. FaST und RationaleRL haben Vorteile hinsichtlich Vielfalt und Neuartigkeit und beide Methoden nutzen Vorwissen. Sowohl REINVENT als auch MOLRL beginnen mit zufälligen Molekülen, die weit vom Trainingsbereich des ML-Klassifikators entfernt sein können.MOLRL erreicht immer noch eine vergleichbare Neuheit wie RationaleRL und erzielt die höchste Erfolgsquote.
Die Verwendung von Vorwissen als Ausgangspunkt kann gewisse Vorteile haben, kann aber auch den Neuartigkeitsgrad und die Fähigkeit des Algorithmus, neue Skelette zu entdecken, einschränken. Darüber hinaus ist die Anwendbarkeit solcher Methoden begrenzt, wenn kein Vorwissen vorhanden ist, beispielsweise bei der Untersuchung unerforschter Ziele.
Neben Aufgaben zur Mehrzieloptimierung besteht ein gängiger Ansatz in der Arzneimittelforschung darin, ein chemisches Gerüst zu identifizieren, das bekanntermaßen an ein bestimmtes Ziel oder eine bestimmte Zielklasse bindet, und dieses als Ausgangspunkt für das chemische Design und die Optimierung zu verwenden. Daher verifiziert das Papier weiter die Fähigkeit von MOLRL, multiobjektive Eigenschaften zu optimieren und gleichzeitig das angegebene Molekülskelett zu erhalten. Wie in der folgenden Tabelle gezeigt,Bei der Optimierung von Molekülen mit einem Aminopyrimidin-Rückgrat erreichte MOLRL eine Erfolgsrate von 100%.
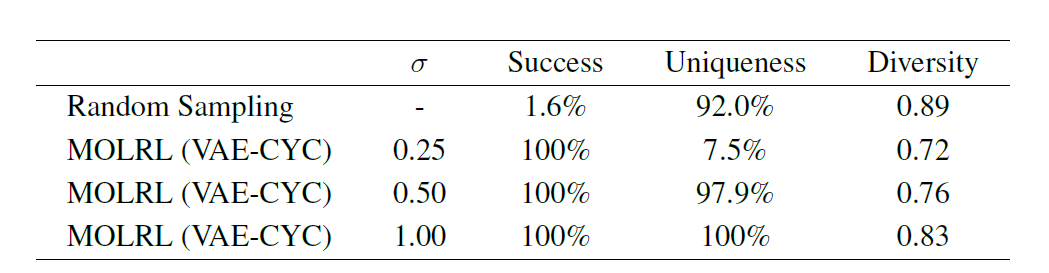
Zusammenfassend lässt sich sagen, dass MOLRL bei einer Vielzahl von Aufgaben im Vergleich zu bestehenden Methoden eine überlegene oder konkurrenzfähige Leistung zeigt.Insbesondere bei der gezielten Molekülgenerierung und Multiparameteroptimierung.
KI ist ein wichtiger Schritt zur Verbesserung der Effizienz der Arzneimittelforschung
Wie viele Ressourcen werden für die Entwicklung eines neuen Medikaments benötigt? In der Pharmaindustrie gilt die berühmte „Doppel-Zehn-Regel“, die besagt, dass es zehn Jahre und eine Milliarde Dollar dauert, bis ein neues Medikament entdeckt und auf den Markt gebracht wird. Laut dem jüngsten Bericht von Deloitte betragen die durchschnittlichen Kosten für die erfolgreiche Markteinführung eines neuen Medikaments für die weltweit führenden Pharmaunternehmen unter Berücksichtigung der Kosten fehlgeschlagener klinischer StudienEs ist von 1,188 Milliarden US-Dollar im Jahr 2010 auf 2,284 Milliarden US-Dollar im Jahr 2022 gestiegen.
Ein entscheidender Schritt bei der Arzneimittelforschung besteht darin, eine Reihe von Kandidatenmolekülen für computergestützte Studien oder die Synthese und Charakterisierung zu finden. Dies ist eine schwierige Aufgabe, da der chemische Raum potenzieller Moleküle riesig ist und ein extrem hoher Aufwand für Versuch und Irrtum erforderlich ist. Heutzutage können künstliche Intelligenz und maschinelles Lernen die Effizienz dieses Schritts effektiv verbessern.
31. Oktober 2023Novartis Institutes for BioMedical Research und Microsoft Research Center for Scientific Intelligence haben zusammengearbeitet, umEine Forschungsarbeit mit dem Titel „Extracting medicinal chemistry intuition via preference machine learning“ wurde in Nature Communications veröffentlicht.
Die Forscher baten 35 Medizinalchemiker, aus 5.000 Molekülpaaren ihr bevorzugtes Molekül auszuwählen, nutzten ihre Antworten, um ein Ranglistenspiel zu erstellen, mit dem ein maschinelles Lernmodell trainiert wurde, und baten das Modell dann, die Moleküle zu bewerten. Diese Bewertung ist weitgehend unbeeinflusst von anderen Eigenschaften, die früher für das Feld charakteristisch waren, da sie auf jahrelangem, in der Branche angesammeltem Wissen beruht.
Das Modell kann das kollektive Wissen, das professionelle Chemiker bei ihrer Arbeit angesammelt haben (oft als „chemische Intuition“ bezeichnet), teilweise reproduzieren und so die zukünftige Arzneimittelentwicklung effizienter gestalten.
Im März 2024 veröffentlichte Insilico Medicine, ein führendes KI-Pharmaunternehmen, eine wissenschaftliche Forschungsarbeit in Nature Biotechnology, in der der Einsatz einer künstlichen Intelligenzplattform zur Entdeckung des neuen Zielmoleküls TNIK zur Behandlung von IPF sowie der anschließende Prozess der Verwendung einer Plattform für generative Chemie zur Entwicklung des Moleküls ISM001-055 detailliert beschrieben werden.
ISM001-055 ist der weltweit erste niedermolekulare Inhibitor.Zielgerichtete Behandlung von TNIK (Traf2/NCK-interagierende Kinase) zur Behandlung der idiopathischen Lungenfibrose (IPF). Laut Insilicon Valley Silicon kann generative KI die Effizienz von Forschung und Entwicklung erheblich steigern, die Kosten senken und die Erfolgsquote in den frühen Phasen der Forschung und Entwicklung erhöhen. Am Beispiel der Moleküle gegen idiopathische Lungenfibrose, von der frühen Zielfindung bis zur Identifizierung präklinischer Kandidatenverbindungen,Es dauerte nur 18 Monate und es wurden 2,6 Millionen Dollar in Forschung und Entwicklung investiert.
Einem Forschungsbericht von Fortune Business Insights zufolge beträgt das weltweite Marktvolumen für künstliche Intelligenz in der Arzneimittelforschung im Jahr 2022 3 Milliarden US-Dollar und dürfte von 3,54 Milliarden US-Dollar im Jahr 2023 auf 7,94 Milliarden US-Dollar im Jahr 2030 wachsen, was einer durchschnittlichen jährlichen Wachstumsrate von 12,21 % entspricht. KI-Technologie hat in Zukunft großes Potenzial, Veränderungen in der Pharmaindustrie voranzutreiben.
Quellen:
1.https://mp.weixin.qq.com/s/OL7TJQcUE-ubhUDyc7GBzQ
2.https://www.thepaper.cn/newsDetail_forward_29097303
3.https://news.bioon.com/article/6127e7234091.html
4.https://bydrug.pharmcube.com/news/detail/49720140c1e9d57ac3c7cfe20ef7f8be
5.https://mp.weixin.qq.com/s/UGAXWMhPlSg2hFnI5ghr1w









