Command Palette
Search for a command to run...
LeCun Weitergeleitet, UC Berkeley Et al. Schlug Eine Multimodale Proteingenerierungsmethode PLAID Vor, Die Sequenzen Und All-Atom-Proteinstrukturen Gleichzeitig Generiert

In den vergangenen Jahren haben Wissenschaftler die Struktur und Zusammensetzung von Proteinen weiter erforscht, um den „Code des Lebens“ besser zu entschlüsseln.Die Funktion eines Proteins wird durch seine Struktur bestimmt, einschließlich der Identität und Position der Seitenketten- und Hauptkettenatome und ihrer biophysikalischen Eigenschaften, die zusammen als allatomare Struktur bezeichnet werden.Um jedoch zu bestimmen, wo die Seitenkettenatome platziert werden sollen, muss man zunächst die Sequenz kennen. Daher kann die Generierung einer Allatomstruktur als multimodales Problem betrachtet werden, das die gleichzeitige Generierung von Sequenz und Struktur erfordert.
Allerdings behandeln bestehende Methoden zur Generierung von Proteinstrukturen und -sequenzen Sequenz und Struktur normalerweise als unabhängige Modi. Methoden zur Strukturgenerierung generieren normalerweise nur Hauptkettenatome. Methoden, die auf ein All-Atom-Design abzielen, erfordern normalerweise die Verwendung externer Modelle, um zwischen Strukturvorhersage und Antifaltungsschritten usw. zu wechseln.
Um diese Herausforderungen zu bewältigen, hat ein Forschungsteam der University of California, Berkeley (UC Berkeley), Microsoft Research und Genentech eine multimodale Methode zur Proteingenerierung namens PLAID (Protein Latent Induced Diffusion) vorgeschlagen, die eine multimodale Generierung durch die Zuordnung von reichhaltigeren Datenmodalitäten (wie Sequenzen) zu knapperen Modalitäten (wie Kristallstrukturen) erreichen kann.Um den Ansatz zu validieren, führten die Forscher Experimente mit 2.219 Funktionen aus der Genontologie und 3.617 Organismen im gesamten Stammbaum des Lebens durch.Obwohl während des Trainings keine strukturellen Eingaben verwendet werden, weisen die generierten Beispiele eine starke strukturelle Qualität und Konsistenz auf.
Die zugehörige Forschung trägt den Titel „Generating All-Atom Protein Structure from Sequence-Only Training Data“ und wurde bei der Top-Konferenz ICLR 2025 eingereicht. „KI-Pate“ Yang Likun hat diesen Erfolg auch auf der sozialen Plattform erneut gepostet.
Open-Source-Adresse des PLAID-Projekts:
http://github.com/amyxlu/plaid
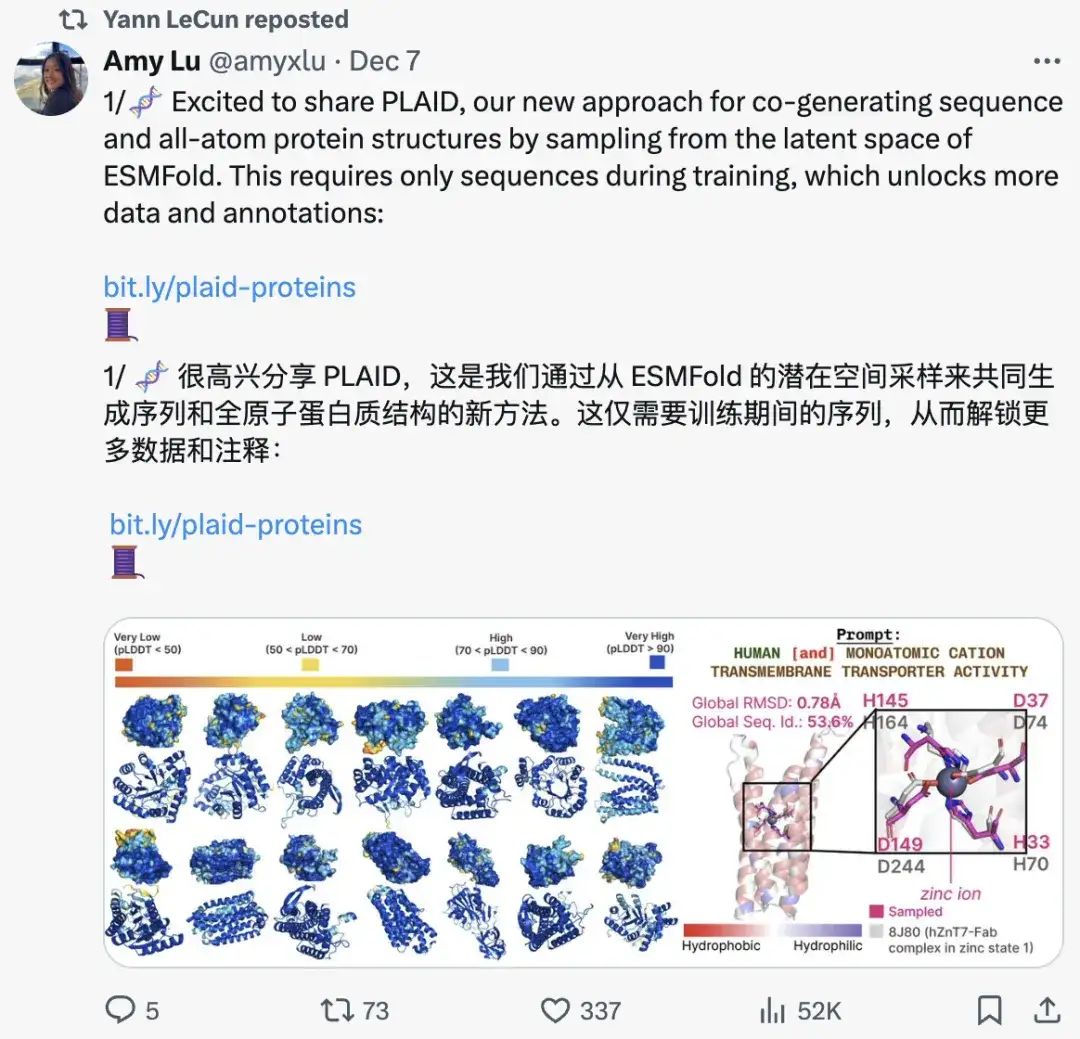
Forschungshighlights:
* Mit Fokus auf das große Proteinsprachenmodell ESMFold und die Generierung reiner Atomstrukturen schlugen die Forscher ein steuerbares Diffusionsmodell vor, das gleichzeitig Sequenzen und rein atomare Proteinstrukturen generieren kann und während des Trainings lediglich die Sequenzeingabe erfordert.
* Der Ansatz nutzt Strukturinformationen, die in vorab trainierten Gewichten kodiert sind, statt in Trainingsdaten und erhöht die Verfügbarkeit von Sequenzanmerkungen für eine kontrollierbare Generierung.
* Obwohl in diesem Dokument das ESMFold-Modell verwendet wird, kann die Methode auf jedes Prognosemodell angewendet werden.
Papieradresse:
https://www.biorxiv.org/content/10.1101/2024.12.02.626353v1
Das Open-Source-Projekt „awesome-ai4s“ vereint mehr als 100 AI4S-Papierinterpretationen und stellt umfangreiche Datensätze und Tools bereit:
https://github.com/hyperai/awesome-ai4s
Ein kurzer Überblick über die Forschungshighlights
Datensatz
Die Forscher verwendeten die Pfam-Datenbank in der Version vom September 2023, die 57.595.205 Sequenzen und 20.795 Familien enthält. PLAID ist vollständig kompatibel mit größeren Sequenzdatenbanken wie UniRef oder BFD (ca. 2 Milliarden Sequenzen). In dieser Studie wurde jedoch die Verwendung von Pfam gewählt, da dessen Sequenzdomäne mehr strukturelle und funktionelle Tags enthält, was die Auswertung der generierten Proben durch Computersimulationen erleichtert. Darüber hinaus behielten die Forscher etwa 15% an Daten zur Überprüfung.
Die UniRef-Codes für Organismen, von denen die Pfam-Domänen abgeleitet wurden, sind in der Datei Pfam-A.fasta verfügbar, die auf dem Pfam-FTP-Server bereitgestellt wird. Die Forscher analysierten alle einzigartigen Organismen im Datensatz, fanden insgesamt 3.617 verschiedene Organismen und führten dann Experimente mit diesen Organismen durch, um die Wirksamkeit der PLAID-Methode zu überprüfen.
Modellarchitektur
PLAID ist ein neues Paradigma für die multimodale, kontrollierbare Erzeugung von Proteinen durch Diffusion im latenten Raum von Vorhersagemodellen.Die Methodenübersicht ist in der folgenden Abbildung dargestellt. Kurz gesagt, es ist in 4 Schritte unterteilt:
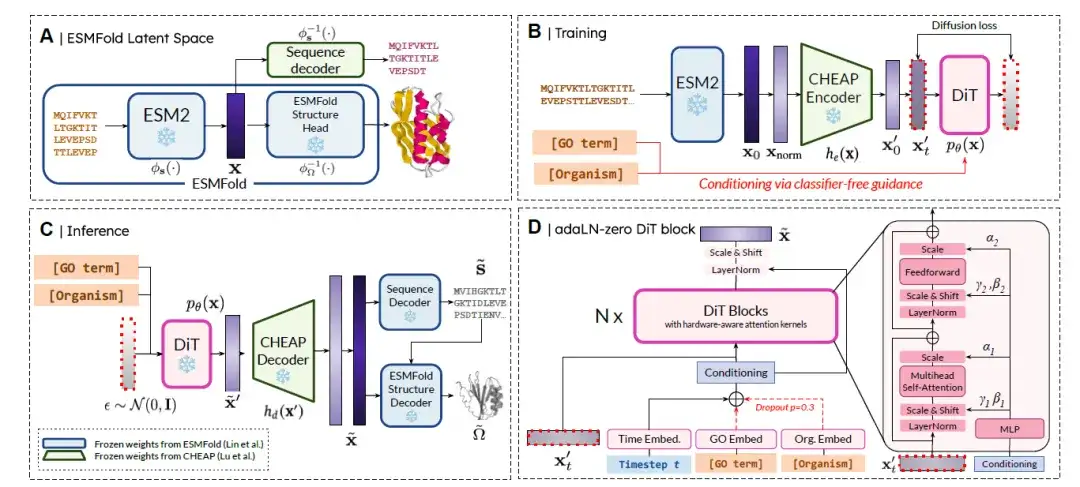
(A) ESMFold Latent Space:Der latente Raum p(x) stellt die gemeinsame Einbettung von Sequenz und Struktur dar.
(B) Training zur Potenzialdiffusion:Das Ziel besteht darin, aus pθ(x) zu lernen und Stichproben zu ziehen, indem man der Diffusionsformel folgt. Um die Lerneffizienz zu verbessern, verwenden die Forscher den CHEAP-Encoder he(·), um die komprimierte Einbettung x′ = he(x) zu erhalten, sodass das Diffusionsziel die Stichprobennahme aus pθ(he(x)) wird.
(C) Schlussfolgerung:Um zum Zeitpunkt der Inferenz sowohl die Sequenz als auch die Struktur zu erfassen, verwenden wir das trainierte Modell, um ˜x′ ∼ pθ(x′) abzutasten und es dann mit dem CHEAP-Decoder zu dekomprimieren, um ˜x = hd(˜x′) zu erhalten. Die Einbettung wird von einem in CHEAP trainierten Frozen Sequence Decoder in die entsprechende Aminosäuresequenz dekodiert. Die Restidentitätssequenz und ˜x werden als Eingabe für einen in ESMFold trainierten Frozen-Structure-Decoder verwendet, um die Gesamtatomstruktur zu erhalten.
(D) DiT-Blockarchitektur:Die Forscher verwendeten die Diffused Transformer (DiT)-Architektur in Kombination mit dem adaLN-zero DiT-Block, um die bedingten Informationen zu fusionieren. Funktionale (d. h. GO-Begriffe) und Organismusklassenbezeichnungen wurden ohne Klassifikatorführung eingebettet.
Studienergebnisse
Die Forscher führten eine Strukturqualitäts- und Diversitätsanalyse verschiedener Proteinlängen durch. Die Ergebnisse sind in der folgenden Abbildung dargestellt.Die nativen Protein- und PLAID-generierten Proben weisen bei unterschiedlichen Längen konsistente Metriken auf.Bei ProteinGenerator und Protpardelle kam es bei bestimmten Längen zu einem Moduskollaps, während bei Multiflow bei längeren Sequenzen eine verringerte Diversität zu beobachten war.
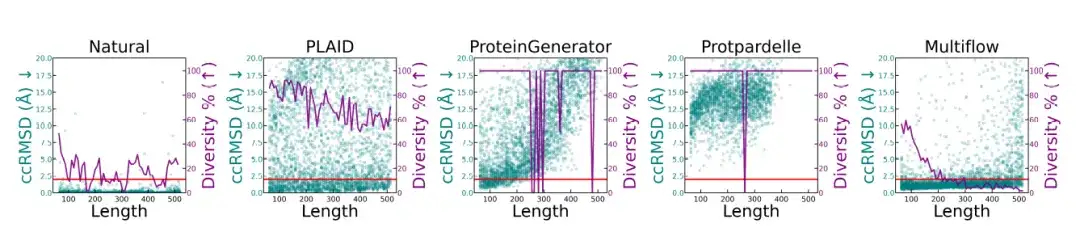
* Diese Abbildung vergleicht natürliche Proteine und verschiedene Erzeugungsmethoden und zeigt die strukturelle Qualität (ccRMSD, cyanfarbene Punkte) und Diversität (violette Linie, gemessen als Anteil einzigartiger Strukturcluster in der Gesamtprobe) von Proteinen unterschiedlicher Länge (64–512 Reste). Die rote Linie liegt bei 2 Å und zeigt den Designschwellenwert an.)
Darüber hinaus wurde im Vergleich zur BasismethodeDie durch PLAID erzeugte Sekundärstrukturvielfalt ähnelt stärker der Verteilung nativer Proteine.Wie in der folgenden Abbildung gezeigt: ProteinGenerator, Protpardelle und Multiflow weisen Abweichungen in ihren Sekundärstrukturverteilungen auf, und vorhandene Modelle zur Generierung von Proteinstrukturen haben normalerweise Schwierigkeiten, Proben mit hohem β-Faltblatt-Gehalt zu generieren.
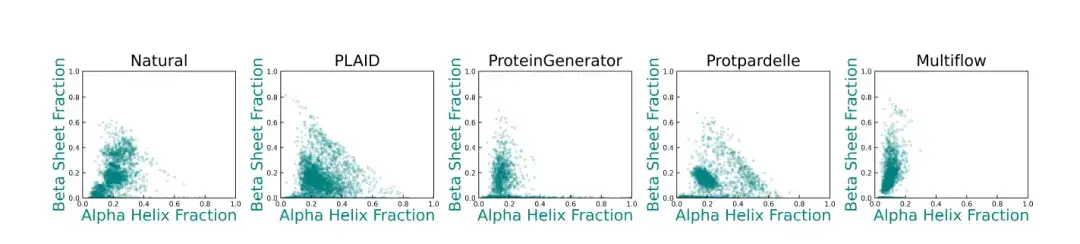
* Die Abbildung zeigt die Verteilung des α-Helix- und β-Faltblatt-Gehalts von natürlichen Proteinen und von Proteinstrukturen, die mit verschiedenen Methoden erzeugt wurden. Jeder Punkt stellt eine Struktur dar und seine Koordinaten stellen den Anteil der α-helikalen Reste (x-Achse) und den Anteil der β-Faltblattreste (y-Achse) dar.
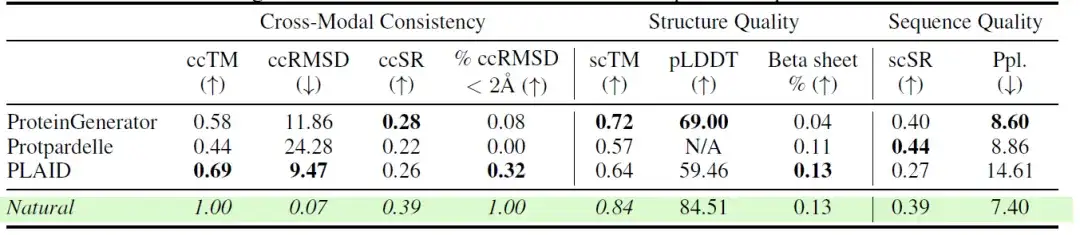
Die Forscher verglichen außerdem die Leistung verschiedener Modelle anhand mehrerer Konsistenz- und Qualitätsmetriken bei der Aufgabe der All-Atom-Proteingenerierung. Die Ergebnisse sind in der folgenden Tabelle dargestellt:Die von PLAID generierten Proben zeigen eine hohe kreuzmodale Konsistenz zwischen Sequenz und Struktur.
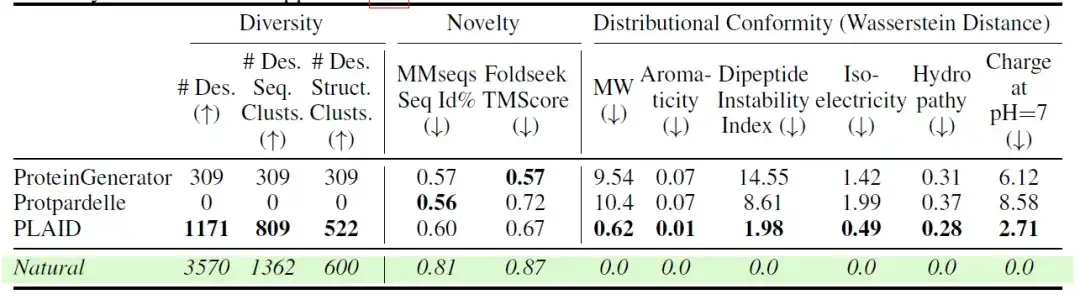
Darüber hinaus bewerteten die Forscher die Vielfalt, Neuartigkeit und Natürlichkeit verschiedener Modelle. Die Ergebnisse sind in der folgenden Tabelle dargestellt:Unter den All-Atom-Modellen hat PLAID sowohl im Sequenz- als auch im Strukturraum die einzigartigsten und am besten gestalteten Proben generiert.
Es ist hervorzuheben, dass PLAID problemlos auf viele nachgelagerte Funktionen erweitert werden kann und nicht auf ESMFold beschränkt ist, sondern auf jedes Vorhersagemodell angewendet werden kann.
KI eröffnet neue Wege in der Proteinforschung
Diffusionstransformatoren werden zunehmend im biologischen Bereich eingesetzt
In diesem Artikel wird erwähnt, dass die Forscher während des Modellerstellungsprozesses den Diffusion Transformer (DiT) zur Durchführung von Rauschunterdrückungsaufgaben verwendeten.
Das Grundprinzip von DiT besteht darin, die Transformer-Architektur auf das Diffusionsmodell anzuwenden. Diffusionsmodelle verfälschen die Originaldaten normalerweise durch schrittweises Hinzufügen von Rauschen und stellen diese Daten dann durch Modelllernen wieder her. DiT verbessert die generative Fähigkeit des Modells durch die Einführung von Transformer-Blöcken (wie adaptive Schichtnormalisierung, Cross Attention usw.) in das Diffusionsmodell.
In den letzten Jahren hat DiT erhebliche Fortschritte im Bereich der Bild- und Videogenerierung erzielt. Die Hauptarchitektur hochmoderner Generationsmodelle wie Sora ist DiT.Im Bereich der Biomedizin wird der Einsatz von Diffusionstransformatoren immer umfangreicher. Es kann Forschern dabei helfen, potenzielle Arzneimittelmoleküle schnell zu prüfen und ihre biologische Aktivität vorherzusagen. Es kann auch bei komplexen Aufgaben wie der Gensequenzanalyse und der Vorhersage von Proteinstrukturen hilfreich sein und stellt somit ein leistungsstarkes Werkzeug für die biowissenschaftliche Forschung dar.Am Beispiel der Protein-Rauschunterdrückung kann DiT komplexe Sequenz-Struktur-Beziehungen erfassen. Das heißt, durch den globalen Selbstaufmerksamkeitsmechanismus von Transformer kann dieser die komplexe interaktive Beziehung zwischen Proteinsequenz und -struktur effektiv modellieren und dann den inversen Prozess des Diffusionsmodells verwenden, um den entrauschten latenten Vektor bei jedem Zeitschritt vorherzusagen und so schrittweise die Struktur und Sequenz des Proteins aus dem Rauschen wiederherzustellen.
Speziell für dieses Dokument bietet DiT flexiblere Optionen zur Feinabstimmung, um mit gemischten Eingabemodalitäten umgehen zu können, insbesondere da Modelle zur Vorhersage von Proteinstrukturen beginnen, Nukleinsäuren und Ligandenkomplexe kleiner Moleküle zu integrieren. Darüber hinaus wird bei diesem Ansatz die Transformer-Schulungsinfrastruktur besser genutzt.
In frühen Experimenten stellten die Forscher außerdem fest, dass die Zuweisung des verfügbaren Speichers an größere DiT-Modelle effizienter war als die Verwendung der dreieckigen Selbstaufmerksamkeit. Es verwendet das von xFormers implementierte Trainingsmodell für Optimierungsalgorithmen und erreichte im Benchmarktest der Inferenzphase eine Geschwindigkeitsverbesserung von 55,8% und eine Reduzierung der GPU-Speichernutzung um 15,6%.
Maschinelles Lernen macht maßgeschneiderte Proteine zu einem „Traum, der wahr wird“
Man kann sagen, dass die oben erwähnte Forschung der UC Berkeley ein weiterer wichtiger Schritt vorwärts in der Proteinanpassung ist. Wir wissen, dass Proteine typischerweise aus 20 verschiedenen Aminosäuren bestehen, die als Bausteine des Lebens gelten können.Aufgrund der extrem komplexen Struktur war es für Wissenschaftler vor Jahrzehnten noch ein Wunschtraum, die dreidimensionale Struktur von Proteinen vorherzusagen und neue Proteine für den menschlichen Gebrauch zu entwickeln. Die rasanten Fortschritte des maschinellen Lernens in den letzten Jahren haben den Traum von der Entwicklung maßgeschneiderter Proteine jedoch allmählich Wirklichkeit werden lassen.
Neben dem bekannten AlphaFold sind auch einige Forschungsfortschritte bemerkenswert:
Im November 2024 entwickelte ein Team des Argonne National Laboratory des US-Energieministeriums erfolgreich ein innovatives Computer-Framework namens MProt-DPO.Das Framework kombiniert künstliche Intelligenztechnologie mit den weltbesten Supercomputern und läutet eine neue Ära im Proteindesign ein. Am Beispiel von MProt-DPO haben Wissenschaftler einen neuen Enzymtyp entwickelt, der unter bestimmten Bedingungen chemische Reaktionen effizient katalysieren kann. Im Vergleich zu früheren Designmethoden ist die Effizienz der neuen Enzymreaktion um fast 30% verbessert, was nicht nur den experimentellen Fortschritt beschleunigt, sondern auch mehr Möglichkeiten für industrielle Anwendungen bietet. Darüber hinaus eröffnet die erfolgreiche Anwendung von MProt-DPO auch neue Ideen für das Design antiviraler Proteine. Die entsprechenden Forschungsergebnisse wurden in der IEEE Computer Society unter dem Titel „MProt-DPO: Breaking the ExaFLOPS Barrier for Multimodal Protein Design Workflows with Direct Preference Optimization“ veröffentlicht.
Papieradresse:
https://www.computer.org/csdl/proceedings-article/sc/2024/529100a074/21HUV88n1F6
Proteintaschen sind Stellen auf Proteinen, die für die Bindung an bestimmte Moleküle geeignet sind. Das Proteintaschendesign ist eine der wichtigsten Methoden im Prozess der individuellen Anpassung von Proteinen. Im Dezember 2024 entwickelten die University of Science and Technology of China und ihre Mitarbeiter den Deep-Generation-Algorithmus PocketGen.Proteintaschensequenzen und -strukturen können basierend auf dem Proteingerüst und gebundenen kleinen Molekülen erzeugt werden. Experimente zeigen, dass Indikatoren wie die Affinität und strukturelle Rationalität des PocketGen-Modells traditionelle Methoden übertreffen und auch die Rechenleistung erheblich verbessert wird. Die entsprechenden Forschungsergebnisse wurden in Nature Machine Intelligence unter dem Titel „Efficient generation of protein pockets with PocketGen“ veröffentlicht.
Papieradresse:
https://www.nature.com/articles/s42256-024-00920-9

Ich bin überzeugt, dass die Menschen in Zukunft durch die weitere Anwendung künstlicher Intelligenz im Proteinbereich ein tieferes Verständnis für die Geheimnisse der räumlichen Struktur von Proteinen erlangen werden.
Quellen:
1.https://www.biorxiv.org/content/10.1101/2024.12.02.626353v1
2.https://mp.weixin.qq.com/s/_5_L7bvl-vHtls8gBbfSmQ
3.https://mp.weixin.qq.com/s/sfrm2rj_8kH0JA2vu4NmTw
4.http://www.news.cn/globe/20241014/f7137840e56340f081f9eb819d87ba40/c.html
5.http://www.bfse.cas.cn/yjjz/202412/t20241212_5042432.html
6.https://www.sohu.com/a/826241274_12








